
Large-scale genome analysis of bovine commensal Escherichia coli reveals that bovine-adapted E. coli lineages are serving as evolutionary sources of the emergence of human intestinal pathogenic strains
- Yoko Arimizu1,2,
- Yumi Kirino3,
- Mitsuhiko P. Sato1,
- Koichi Uno4,
- Toshio Sato4,
- Yasuhiro Gotoh1,
- Frédéric Auvray5,
- Hubert Brugere5,
- Eric Oswald5,6,
- Jacques G. Mainil7,
- Kelly S. Anklam8,
- Dörte Döpfer8,
- Shuji Yoshino9,
- Tadasuke Ooka10,
- Yasuhiro Tanizawa11,
- Yasukazu Nakamura11,
- Atsushi Iguchi12,
- Tomoko Morita-Ishihara13,
- Makoto Ohnishi13,
- Koichi Akashi2,
- Tetsuya Hayashi1 and
- Yoshitoshi Ogura1
- 1Department of Bacteriology, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan;
- 2Department of Medicine and Biosystemic Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan;
- 3Laboratory of Veterinary Radiology, Department of Veterinary Science, Faculty of Agriculture, University of Miyazaki, Miyazaki 889-2192, Japan;
- 4Japan Microbiological Laboratory, Sendai, Miyagi 983-0034, Japan;
- 5IRSD, Université de Toulouse, INSERM, INRA, ENVT, UPS, 31300 Toulouse, France;
- 6CHU de Toulouse, Hôpital Purpan, 31300 Toulouse, France;
- 7Bacteriology, Department of Infectious Diseases, Faculty of Veterinary Medicine and Institute for Fundamental and Applied Research in Animal Health (FARAH), University of Liège, 4000 Liège, Belgium;
- 8Department of Medical Sciences, School of Veterinary Medicine, University of Wisconsin, Madison, Wisconsin 53705, USA;
- 9Department of Microbiology, Miyazaki Prefectural Institute for Public Health and Environment, Miyazaki 889-2155, Japan;
- 10Department of Microbiology, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima 890-8520, Japan;
- 11Center for Information Biology, National Institute of Genetics, Research Organization of Information and Systems, Mishima, Shizuoka 411-8540, Japan;
- 12Department of Animal and Grassland Sciences, Faculty of Agriculture, University of Miyazaki, Miyazaki 889-2192, Japan;
- 13Department of Bacteriology I, National Institute of Infectious Diseases, Tokyo 162-8640, Japan
Abstract
How pathogens evolve their virulence to humans in nature is a scientific issue of great medical and biological importance. Shiga toxin (Stx)–producing Escherichia coli (STEC) and enteropathogenic E. coli (EPEC) are the major foodborne pathogens that can cause hemolytic uremic syndrome and infantile diarrhea, respectively. The locus of enterocyte effacement (LEE)–encoded type 3 secretion system (T3SS) is the major virulence determinant of EPEC and is also possessed by major STEC lineages. Cattle are thought to be the primary reservoir of STEC and EPEC. However, genome sequences of bovine commensal E. coli are limited, and the emerging process of STEC and EPEC is largely unknown. Here, we performed a large-scale genomic comparison of bovine commensal E. coli with human commensal and clinical strains, including EPEC and STEC, at a global level. The analyses identified two distinct lineages, in which bovine and human commensal strains are enriched, respectively, and revealed that STEC and EPEC strains have emerged in multiple sublineages of the bovine-associated lineage. In addition to the bovine-associated lineage-specific genes, including fimbriae, capsule, and nutrition utilization genes, specific virulence gene communities have been accumulated in stx- and LEE-positive strains, respectively, with notable overlaps of community members. Functional associations of these genes probably confer benefits to these E. coli strains in inhabiting and/or adapting to the bovine intestinal environment and drive their evolution to highly virulent human pathogens under the bovine-adapted genetic background. Our data highlight the importance of large-scale genome sequencing of animal strains in the studies of zoonotic pathogens.
Escherichia coli are commensal intestinal inhabitants of a wide range of vertebrates. Several types of strains, however, cause diverse intestinal and extraintestinal diseases in humans by means of individually acquired virulence factors (Dobrindt 2005). Shiga toxin (Stx)–producing E. coli (STEC) is a major cause of gastrointestinal illness and often causes serious diseases, including hemorrhagic colitis (HC) and hemolytic uremic syndrome (HUS) (Karch et al. 2005). Stxs are divided into two major groups, Stx1 and Stx2, and both are encoded by lysogenic bacteriophages. Although Stxs are the key factor for the development of both HC and HUS, the major STECs, such as O157:H7, have acquired many other virulence determinants through horizontal gene transfer (Hayashi et al. 2001; Tobe et al. 2006). These include the locus of enterocyte effacement (LEE) pathogenicity island that encodes a type 3 secretion system (T3SS) and several effectors, more than 30 phage-encoded non-LEE effectors, and several plasmid-encoded virulence factors. The LEE-encoded T3SS enables the bacteria to induce attaching and effacing (A/E) lesions, which are characterized by the effacement of the brush border microvilli and intimate bacterial attachment to intestinal epithelial cells (Karch et al. 2005).
Many different O:H serotypes of strains have been identified in STEC infections (Croxen et al. 2013). O157:H7 and the major non-O157 STECs (O26:H11, O111:H8, and O103:H2) belong to different phylogenetic lineages but share a similar set of virulence determinants (Reid et al. 2000; Ogura et al. 2009). Each STEC lineage appears to have independently acquired phages and plasmids that carry a similar virulence gene set, including the LEE, non-LEE effectors, and plasmid-encoding virulence factors (Ogura et al. 2009). LEE-negative STEC strains have occasionally caused HC and HUS, but such strains often carry a virulence determinant alternative to the LEE (Krause et al. 2018).
Enteropathogenic E. coli (EPEC), which are important pathogens responsible for diarrhea particularly in young children, also possess the LEE but do not produce Stx (Croxen et al. 2013). It has been recently shown that there is a great phylogenetic and genomic diversity among EPEC isolates (Hazen et al. 2016), and EPEC has emerged on multiple occasions by acquiring various LEE variants (Ingle et al. 2016a). Ruminant animals, especially cattle, are thought to be the major natural reservoir of STEC and EPEC (Beutin et al. 1993; Holland et al. 1999; Kolenda et al. 2015). However, it is largely unknown how STEC and EPEC emerged in the environment. This is at least partly because E. coli genome sequencing efforts have been highly biased to human pathogenic strains. Here, to elucidate the evolutionary pathway of STEC and EPEC, we performed a large-scale genome analysis of bovine commensal E. coli and compare their genomes with those of human commensal E. coli and clinical STEC, EPEC, and extraintestinal pathogenic E. coli (ExPEC).
Results
Phylogenetic analyses of bovine and human commensal E. coli and clinical isolates
In total, 575 bovine and 362 human commensal E. coli were analyzed in this study (Table 1). In this study, we defined E. coli strains isolated from healthy cattle and humans as bovine and human commensal E. coli, respectively. Therefore, these “commensal strains” may or may not contain canonical E. coli virulence factors. Human clinical STEC/EPEC (n = 86) and ExPEC (n = 111) isolates, which had clear indications that caused diseases in humans, were also included for comparison (for strain selection strategies, see Methods). In general, it is thought that ExPECs colonize large intestine in healthy hosts without causing diseases but can cause various extraintestinal diseases depending on host physiological conditions (Vila et al. 2016). Of the 1134 strains analyzed, 884 were sequenced in this study and 250 were from public databases (for details, see Supplemental Tables S1, S2, respectively). The strains were isolated in various geographic regions (21 countries, six continents) (Supplemental Fig. S1). A total of 227 (∼40%) of the bovine commensal isolates and four of the human commensal isolates were stx and/or LEE positive (Table 1).
Strains used in this study
We first constructed a neighbor-joining (NJ) tree based on seven housekeeping genes of the 937 commensal strains and 34 completely sequenced E. coli reference strains (Fig. 1A) with E. fergusonii and cryptic Escherichia clade 1 strains as outgroups. The NJ tree clearly showed that most bovine commensal strains (549/575) belonged to a large monophyletic lineage (referred to as bovine-associated lineage) that is distinct from a human strain–dominated lineage (referred to as human-associated lineage). E. coli strains have been historically grouped into seven major phylogroups (A, B1, B2, C, D, E, and F). Although phylogroups determined by in silico PCR analysis of four marker genes (Clermont et al. 2013) did not fully correspond to the phylogenetic relationship between the strains in the NJ tree, the bovine-associated lineage included strains belonging to phylogroups A, B1, C, D, and E, and the human-associated lineage included B2, D, and F strains.

Phylogenetic relationship between human and bovine commensal E. coli and distribution of stx- and LEE-positive isolates. (A) The NJ tree of 937 bovine and human commensal isolates with 36 completely sequenced reference strains based on the concatenated nucleotide sequences of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA). The phylogeny was rooted using Escherichia fergusonii strain ATCC35469 and cryptic Escherichia clade 1 strain TW15838. Sources of sequences and phylogroups of strains are shown in the NJ tree. Pathotypes of reference strains are indicated by differently colored characters. (B) The core gene–based ML tree of 937 bovine and human commensal isolates with 197 human clinical isolates. Cryptic Escherichia clade I strain TW15838 was included as an outgroup. The tree was constructed based on 247,627 SNPs located on 1755 core genes. Country, BAPS group, phylogroup, ST, host and pathotype of each strain, and the presence of stx1, stx2, and LEE are shown. Positions of clinical isolates of major STEC serotypes are indicated.
The phylogenetic relationship between bovine and human commensal strains was also evident in a core gene–based maximum likelihood (ML) tree that included 197 human clinical isolates in addition to the 937 commensal strains (Fig. 1B). The bovine- and human-associated lineages were clearly separated by Bayesian analysis of population structure (BAPS) (Cheng et al. 2013). Most bovine (96%; 552 out of 575 strains) and human (73%; 264 out of 362 strains) commensal isolates were grouped into the bovine- and human-associated lineages, respectively, although some portions of human commensal isolates were in the bovine-associated lineage, especially phylogroup A strains. We obtained almost same distribution patterns in the randomization test analyzing equal numbers of bovine and human commensal strains (300 strains randomly selected from each group) (Supplemental Table S3).
Although Japanese isolates were most prevalent in the strain set, strains isolated in various geographic regions were diversely distributed among the Japanese isolates in the core gene tree. In addition, among the 196 O and 53 H serogroups in the E. coli O and H antigen gene database (EcOH database) (Ingle et al. 2016b), 163 and 46 were detected in the strains analyzed, respectively (Supplemental Fig. S2). Furthermore, of the 1134 strains analyzed, 961 were grouped into 372 different sequence types (STs), and 173 were not assigned to any known STs (Fig. 1B; Supplemental Tables S1, S2). These data indicate a great genetic diversity of our strain set. Among the clinical isolates, although most ExPECs (82%) belonged to the human-associated lineage, most STECs and EPECs (79%) belonged to the bovine-associated lineage (Fig. 1B). Ten major STEC serotypes were dispersedly distributed in the bovine-associated lineage. These findings, together with the fact that many stx- and/or LEE-positive strains were included in bovine commensal E. coli, suggests that the origins of STEC and EPEC are bovine commensal E. coli.
Distribution of major STEC and EPEC virulence genes in bovine and human commensal E. coli
We next reconstructed a core gene tree including only commensal isolates and analyzed the distribution of major virulence genes of STEC and EPEC among these strains. As shown in Figure 2A, with two exceptions, all stx-positive strains (n = 144) belonged to the bovine-associated lineage, with a highly scattered distribution. Among the known stx subtypes, stx1a, 2a, and 2c/d were identified in many sublineages (Supplemental Fig. S3; Supplemental Table S4), indicating that the acquisition of phages carrying these stx subtypes frequently occurred in bovine commensal E. coli. Similarly, LEE-positive strains (n = 99) were distributed in many sublineages of the bovine-associated lineage but also in three sublineages in the human-associated lineage (Fig. 2A). Although 53 out of the 99 LEE-positive strains were clustered together in the bovine-associated lineage, they belonged to distinct sublineages and showed diverse serotypes, which include not only two well-known STEC serotypes, O26:H11 (n = 9) and O121:H19 (n = 1), but also O165:H25 (n = 12), O177:H11 (n = 11), O10:H25 (n = 4), O182:H25 (n = 4), and additional 12 serotypes. Seventeen STs were also assigned to the 53 LEE-positive strains.
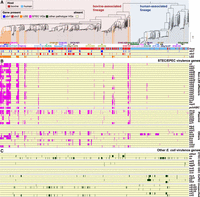
Distribution of virulence genes among 937 bovine and human commensal E. coli strains. (A) The ML tree based on 262,788 SNP sites on 1958 core genes. The tree was rooted by cryptic Escherichia clade I strain TW15838. LEE-positive lineages/strains are highlighted by orange lines. Frequently observed serotypes (more than four strains) and major STEC serotypes are indicated. The presence of stx1, stx2, and LEE in each strain is shown. The presence of other STEC/EPEC virulence genes (B) and virulence genes associated with other E. coli pathotypes (C) is shown, respectively. For the functions and nucleotide sequences of each gene analyzed, see Supplemental Table S7.
Among the 30 known LEE subtypes based on the intimin gene sequence (Ooka et al. 2012), 11 were detected in the commensal E. coli analyzed in this study. The intimin-based phylogeny of LEE-positive strains correlated well with the LEE core gene–based phylogeny (Supplemental Fig. S4A). However, the distributions of LEE subtypes did not follow the phylogeny of the LEE-positive strains inferred by the whole-genome core gene sequences, indicating that the acquisition of the whole LEE element has occurred in many E. coli sublineages (Supplemental Fig. S4B). This supports previous findings that the LEE is transferable between or to E. coli although the LEE itself contains no gene required for horizontal transfer (Deng et al. 2001; Hazen et al. 2013; Ingle et al. 2016a).
Non-LEE effectors and PchABC family regulators (one of the master regulators of the gene expression of LEE and effectors) (Iyoda and Watanabe 2004) were detected only in LEE-positive strains (Figs. 2B, 3A), despite the fact that they are encoded by mobile genetic elements (MGEs), mostly by phages. This indicates that these genes were horizontally acquired and stably maintained in these strains, suggesting that in bovine intestine, there are some selection pressures and mechanisms to stimulate the accumulation of these virulence factors in LEE-positive E. coli strains.
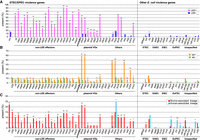
Conservation of STEC/EPEC-related and other E. coli virulence genes in bovine and human commensal isolates. Conservation of the virulence genes of STEC/EPEC and those of other E. coli pathotypes in LEE-positive (n = 104) and LEE-negative (n = 833) strains (A), in stx-positive (n = 146) and stx-negative (n = 791) strains (B), and in the bovine-associated (n = 647) and human-associated (n = 290) lineages (C) are summarized. Statistical analyses were performed using the two-sided Fisher's exact test. Asterisks indicate that the differences were significant after the Bonferroni correction for multiple comparisons: (*) P < 0.05; (**) P < 1 × 10−6.
Comparison of bovine commensal and human clinical LEE-positive isolates
The LEE-positive strains were further analyzed for their phylogenetic distribution and non-LEE effector repertoires. Bovine commensal and human clinical LEE-positive isolates were grouped together on multiple BAPS clades in the whole-genome core gene–based ML tree (Supplemental Fig. S5). Statistically significant differences in effector repertoires were not detected, except for tccP, between the bovine commensal and human clinical strains. Thus, the strains from these two sources were not clearly distinguishable in terms of phylogeny and non-LEE effector repertoires, although much larger scale analyses are required to obtain a complete understanding of this issue. It may also be noteworthy that the bundle-forming pilus (BFP), which mediates localized adherence to epithelial cells by so called “typical EPEC” (Nataro and Kaper 1998; Nougayrede et al. 2003), was absent in all bovine commensal LEE-positive E. coli isolates (Figs. 2B, 3); however, it is known that bfp-negative EPECs also frequently cause human diseases (Ochoa and Contreras 2011). The source(s) of bfp-positive EPEC remains to be elucidated.
Distribution of other STEC/EPEC virulence genes in bovine and human commensal E. coli
In addition to the major virulence genes, various genes suspected to be related to virulence were identified in STEC and EPEC (Dobrindt 2005; Krause et al. 2018). The distribution of these STEC/EPEC virulence genes in our commensal strain set was analyzed. Virulence genes encoded by STEC virulence plasmids, such as pO157 and pO26, accumulated significantly more in LEE-positive strains than in LEE-negative strains (Figs. 2B, 3A). Among these, ehxA and espP were also more frequently detected in stx-positive strains than in stx-negative strains (Figs. 2B, 3B). These data suggest that the coexistence of these plasmid-encoded virulence genes with LEE and/or stx can be an adaptive advantage for E. coli in bovine intestine. Consistent with this presumption, the expression of ehxA is regulated by the LEE-encoded transcriptional regulators, Ler and GrlA (Iyoda et al. 2011). Moreover, among the other STEC/EPEC virulence genes, lpxR, paa, and ureC showed a strong association with LEE (Fig. 3A). A significant association of hes, iha, saa, sab, and subA with stx was also observed (Fig. 3B). These data suggest that these genes are functionally related to LEE or Stx. Such associations may also provide adaptive advantages to E. coli in the bovine intestinal environment.
Distribution of other E. coli virulence genes in bovine and human commensal E. coli
The distribution of virulence genes identified in other E. coli pathotypes was also analyzed. This was performed as E. coli O104:H4 and O80:H2, which are hybrid pathotypes of STEC with enteroaggregative E. coli (EAEC) and ExPEC, respectively, have recently emerged and caused large outbreaks of HUS in Europe (Navarro-Garcia 2014; Soysal et al. 2016). Among the 19 genes analyzed, two showed distributions significantly biased to the bovine-associated lineage (Fig. 3C). Conversely, the distributions of four genes showed significant biases to the human-associated lineage. Contrasting to the STEC/EPEC virulence genes, virulence determinants of other pathotypes showed no significant association with stx (Fig. 3B), suggesting that the emergence of hybrid STEC with other pathotypes may be an accidental event.
Co-occurrence network analysis of virulence genes
Co-occurrence of the virulence genes of STEC/EPEC and other E. coli pathotypes was further analyzed by a network analysis (Fig. 4). This analysis identified seven gene communities (named communities 1–7). As expected, LEE, non-LEE effectors, and pchABC were grouped together with all plasmid virulence genes examined and three other STEC/EPEC virulence genes (lpxR, paa, and ureC) into a large cluster (community 1). stx was grouped into another cluster (community 2), in which two plasmid virulence genes (espP and ehxA) and four other STEC/EPEC virulence genes (hes, ihaA, saa, and subA) appeared frequently. Functional association of virulence genes within each gene community may enhance the niche adaptation of E. coli harboring each community and/or drive the further evolution of virulence potentials of EPEC and STEC, respectively.
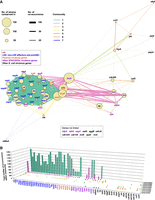
Co-occurrence network analysis of virulence genes. (A) Co-occurrence network analysis of virulence genes in bovine and human commensal isolates. Gene names are colored based on the virulence gene classification indicated in the box. The node sizes represent the number of strains in which each gene was conserved. Seven communities identified are indicated by differently colored edges (connections). Edge widths represent the number of co-occurrences. Only one co-occurrence between genes was excluded from the analysis. (B) Community members and their total numbers of co-occurrence within each community.
It is also of note that communities 1 and 2 were linked through several genes, including ihaA, espP, ehxA, and stx (Fig. 4). LEE-positive STEC strains have more frequently caused severe diseases such as HC and HUS than LEE-negative STEC strains; STEC strains that caused or potentially cause HC and HUS are sometime called enterohemorragic E. coli (EHEC) (Croxen et al. 2013). Functional linkage between these two virulence gene communities may be associated with or stimulate the emergence and dissemination of more virulent “EHEC” strains in the environment.
Larger genome sizes and higher integrase and prophage numbers in stx- and/or LEE-positive E. coli commensal strains
Assuming that phages carrying virulence genes have accumulated in stx- and/or LEE-positive strains, these strains should contain larger genomes than double-negative strains. As expected, although median total scaffold lengths were not apparently different between bovine and human commensal strains, those of stx- and/or LEE-positive commensal strains were significantly longer than those of the double-negative commensal strains (Supplemental Fig. S6A,B; Supplemental Table S5). Consistent with this, the numbers of predicted integrases and prophage regions were higher in stx- and/or LEE-positive strains compared with the double-negative strains (Supplemental Fig. S6D,F; Supplemental Table S5).
Genes specifically present in the bovine or human-associated lineages
Finally, we searched for the genes that are specifically present in the bovine- or human-associated lineages. Among the genes identified in 937 bovine and human commensal E. coli strains, 1697 and 28,885 were assigned as core and accessory genes, respectively. Based on the presence and absence matrix of the pan-genome, 2879 genes were identified as being positively or negatively associated with the bovine-associated lineage (Fig. 5A; Supplemental Table S6).

Bovine- or human-associated lineage-specific gene. (A) A scattered plot of gene conservation in the bovine- and human-associated lineages. Genes that were significantly (positively or negatively) associated with the bovine-associated lineage (Bonferroni P < 0.05) are indicated by blue dots. Among the positively associated (bovine-associated lineage-specific) and negatively associated (human-associated lineage-specific) genes, the top 50 genes with known or predictable functions (Bonferroni P < 1 × 10−135 and P < 1 × 10−141, respectively) are indicated in each group by purple and red dots, respectively. (B) Distribution of the bovine- or human-associated lineage-specific genes in the core gene–based ML tree (the same tree shown in Fig. 2). Presence and absence of each gene are indicated by purple and beige, respectively.
Among the genes with known or predictable functions, the top 50 genes in each group were analyzed in more detail. Bovine-associated lineage-specific genes included 14 genes for the biosynthesis of five different fimbriae (Fig. 5B). We speculate that these are important colonization factors in the bovine intestine. Other notable bovine-associated lineage-specific genes were a set of genes (n = 7) for O-antigen capsule (group 4 capsule) biosynthesis. In E. coli, the O-antigen capsule was shown to be required for colonization in the bovine intestine (Dziva et al. 2004). A set of genes (n = 14) for phenylacetate utilization and xylose and melibiose transport was also bovine-associated lineage-specific, suggesting that the ability to use these nutrient sources is beneficial for E. coli to adapt to bovine intestine. Negatively associated genes (human-associated lineage-specific genes) included the kps genes (n = 5) for the biosynthesis of group 2 and 3 capsules, which are known to be produced mainly by ExPEC (Whitfield 2006). Genes for three iron utilization systems (n = 18) and a multidrug efflux system (n = 5) were also found to be human-associated lineage-specific.
Different phosphotransferase system (PTS) genes for fructose transport were found to be associated with the two E. coli lineages, respectively. Although the origins and acquisition/inheritance histories of these PTS systems are yet to be analyzed, the ability to use fructose may be beneficial for E. coli in intestinal environments of both humans and bovines.
Discussion
Here, we performed a large-scale genomic comparison of bovine commensal E. coli with human commensal and clinical strains, including EPEC, STEC, and ExPEC, and revealed that bovine commensal E. coli strains are phylogenetically distinct from human commensal strains. In our data set, the bovine-associated lineage mainly consisted of strains belonging to phylogroups B1 (72%) and A (23%), and the human-associated lineage mainly consisted of B2 (66%) and D (24/%) strains (Fig. 1). These findings are basically concordant with the previous finding that most bovine commensal isolates belonged to phylogroups B1 and A (Bok et al. 2015; Madoshi et al. 2016; Mercat et al. 2016). As for human commensal isolates, it was reported that B2 strains predominate in people residing in developed countries in the temperate regions of the world (Escobar-Paramo et al. 2004; Skurnik et al. 2008). It has also been reported that phylogroup B2 or D strains predominated (>70%) in the E. coli strains isolated from biopsy samples of human lower intestinal tracts in Australia (Gordon et al. 2015). However, in a recent analysis of E. coli strains isolated from healthy individuals who traveled from the United Kingdom to South Asia (Bevan et al. 2018), not only phylogroups B2 and D strains but also phylogroup A strains were predominant. In another recent study that analyzed E. coli isolates from Tanzanian children under the age of 5, phylogroups A and B1 strains were most frequently isolated (Richter et al. 2018). Although 56 of the 168 phylogroup A strains in our data set were human commensal isolates (Fig. 1), the difference in the proportion of phylogroup A strains between studies may be owing to the difference in geography or host age.
An important outcome of this study is the identification of genes that show biased distribution to the bovine- or human-associated lineage (Fig. 5), which may contribute the adaptation of each lineage to bovine and human intestinal environments. It is noteworthy that several sublineages (corresponding to phylogroups A, D, E, and F) in the bovine- and human-associated lineages, which early separated from others in each lineage, showed a mixed presence/absence pattern of these lineage-specific genes, suggesting that these sublineages, particularly the sublineage comprising phylogroup A strains, which contained both bovine and human commensal strains (Figs. 1, 2), may represent intermediates in the emergence process of bovine- or human-adapted lineage. In the same context, it may be possible to regard phylogroups B1 and B2 as the lineages most adapted to bovine and human intestine, respectively. In fact, B1- and B2-specific genes identified by a gene repertoire comparison focused on B1 and B2 strains (Supplemental Table S9) included not only most of the bovine- and human-associated lineage-specific genes listed in Figure 5 but also many additional genes involved in various cellular functions that may also be required for better adaptation to each host.
Another important finding of this study was that most clinical STEC and EPEC isolates belonged to the multiple sublineages in the bovine-associated lineage and were indistinguishable from stx- and/or LEE-positive bovine commensal E. coli (Fig. 1; Supplemental Fig. S5), indicating that STEC and EPEC strains have emerged on multiple occasions from bovine commensal E. coli. We also present evidence for the specific distribution of non-LEE effectors in LEE-positive strains (Figs. 2, 3), which suggests the presence of a strong selection pressure to accumulate and stably maintain these effectors in LEE-positive strains in the bovine intestinal environment. Other STEC and EPEC virulence genes were also found to be specifically distributed in stx- or LEE-positive strains, suggesting their functional association with Stx or LEE. The network analysis supported functional links of these genes (Stx- or LEE-associated virulence gene communities) and further suggested the presence of a linkage between the two virulence gene communities via several shared community members (Fig. 4). We speculate that the coexistence of these genes is advantageous for the adaptation to the bovine intestinal environment and promotes the further evolution of STEC and EPEC to be more virulent pathogens for humans. In such evolutionary processes, the presence of bacterivorous protozoa that naturally inhabit the bovine intestine, especially in the rectal end, may be one of the possible selection pressures that are exerted, as Stx- and LEE-encoded T3SS have been shown to show antipredation activities against predators (Erken et al. 2013). However, more complete understanding of the processes and driving forces of STEC and EPEC evolution would be required to develop efficient strategies to reduce the current large burden of the emergence and prevalence of these pathogens.
Methods
Bacterial strains and DNA sequencing
We collected 1666 rectal swab samples from healthy adult cattle in various farms in seven prefectures in Japan from 2013–2014. Each sample was incubated in mEC broth (Nissui) without shaking overnight at 42°C and then plated on XM-G agar medium (Nissui). PCR was performed using 1 µL of boiled mEC culture of each sample as a template and stx1-specific, stx2-specific, and eaeA (a marker for LEE)-specific primers as described elsewhere (Ogura et al. 2015). Of the 1666 samples, stx, eaeA, or both were detected in 332 (20%), 162 (10%), or 369 (22%) samples, respectively. After overnight incubation of the XM-G plate at 37°C, 48 colonies were transferred to a 96-well plate, which contained 100 µL of lysogeny broth medium per well, and were incubated overnight at 37°C. The presence of stx1, stx2, and eaeA in each colony was analyzed by PCR using 1 µL of boiled culture as a template and primers as described above. From each sample, one or more clones (if different PCR patterns were shown) were selected and stored as glycerol stock at −80°C. In total, 661 stx- and/or LEE-positive and 411 double-negative isolates were obtained. For sequencing, we randomly selected 298 stx- and/or LEE- positive strains and 227 double-negative strains (565 in total). An additional 40, 45, and 105 commensal E. coli strains were isolated from healthy adult cattle in Belgium, the United States, and France, respectively.
In addition, 331 E. coli strains were isolated from the stools of healthy adult humans in Japan from 2008 to 2015 using XM-G agar medium. These healthy humans included food handlers and workers in daycare centers for children and elders. These workers are required by law to undergo periodic fecal examination. Animal handlers were not included in the examinees. For the isolation of human commensal E. coli, only one colony was picked from a XM-G plate for each stool sample. Furthermore, we collected 103 E. coli strains (ExPEC strains) isolated from blood or urine specimens of patients from various hospitals in Japan. In total, 1149 strains were sequenced in this study.
Genomic DNA was purified from 1 mL of an overnight culture of each strain using a DNeasy blood and tissue kit (Qiagen). Genomic DNA libraries were prepared using the Nextera XT DNA sample preparation kit (Illumina) and sequenced using the Illumina MiSeq platform to generate 300-bp paired-end reads.
The genome sequence information of 52 bovine commensal and 86 human commensal strains and 112 clinical strains (STEC, EPEC, and ExPEC) isolated in various countries was obtained from public databases along with metadata (Supplemental Table S2). All genome data of bovine and human commensal isolates available in the NCBI database (accessed in January 2019) were included in this study. As for clinical STEC and EPEC strains, we first selected the strains, for which necessary metadata including country, host, and clinical significance (disease or symptom) are available, from those in the NCBI database (accessed in August 2017), and then one strain was randomly selected from each serotype (according to the original description). Regarding ExPEC strains, we selected strains with the metadata mentioned above from the strains in three BioProjects (PRJNA269984, PRJEB9927, PRJNA266030) and further selected 26 strains having different serotypes and not closely related to each other (less than five SNP distance).
Assembly and annotation
Genome assembly, scaffolding, and gap-closing of the Illumina sequence reads obtained in this study and from public databases were performed using the Platanus assembler (Kajitani et al. 2014). Original assemblies were used if assembled sequences were publicly available. Annotation was performed with the DNA Data Bank of Japan (DDBJ) Fast Annotation and Submission Tool (DFAST) (Tanizawa et al. 2018).
ST, serotype, and phylogroup determinations and stx and eaeA subtyping
ST determination and stx1 and stx2 subtyping were performed by a read mapping–based strategy using the SRST2 program (maximum 1% divergence) (Inouye et al. 2014). In the public database sequences, for which raw read data were not available, the reads were simulated with the wgsim version 0.3.2 (https://github.com/lh3/wgsim) using the default parameters. In silico serotyping was conducted by BLASTN search (>85% identity and >60% coverage) of scaffold sequences of each strain against the database file EcOH.fasta that is distributed with SRST2. Phylogroup was determined by ClermonTyping (Beghain et al. 2018). Subtypes of eaeA genes were determined by BLASTN search (>96% identity with 96% coverage). Reference sequences of each subtype of stx and eae have been described elsewhere (Ooka et al. 2012; Scheutz et al. 2012).
Phylogenetic analysis
NJ trees based on seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA), the intimin gene (eae), and six LEE core genes (escS, escC, escJ, escV, escN, and cesD2) were constructed by MEGA7 (Kumar et al. 2016) using the Tamura-Nei evolutionary model.
To construct core gene–based phylogenetic trees, the pan-genome analyses for each strain set were performed using Roary (Page et al. 2015). Core genes were defined as genes present in ≥99% of strains with ≥80% nucleotide sequence identity. SNP sites were extracted from the core gene alignment using SNP-sites (Page et al. 2016). After removal of the sites with ≥5% ambiguous base call and gaps, ML phylogenetic trees were constructed using RAxML (Stamatakis 2006) with the GTR-GAMMA model of nucleotide substitution and 500 bootstrap replicates. The ML phylogenetic trees were displayed and annotated using iTOL (Letunic and Bork 2016). Clustering analysis was performed using the hierarchical Bayesian Analysis of Population Structure (hierBAPS) program (Cheng et al. 2013).
Of the strains sequenced in this study, strains with low sequence coverage (<×25) were excluded (n = 54). Strains that were found to belong to cryptic Escherichia lineages or species (n = 35) and those that showed five or less of SNP difference to one of the other strains (n = 176) were also excluded from the analyses. Finally, 884 strains were used for further analyses and are listed in Supplemental Table S1.
Detection of virulence genes
Presence of non-LEE effectors was analyzed by TBLASTN search (>50% identity and >50% coverage) using amino acid sequences as query. Other virulence genes were identified using the SRST2 with the default setting. Amino acid sequences and nucleotide sequences used for the detection of virulence factors are listed in Supplemental Table S7.
Co-occurrence network analysis of virulence genes
To analyze the co-occurrence of virulence genes among bovine and human commensal E. coli strains and visualize it in the network interface, we constructed a pairwise co-occurrence matrix for each gene (Supplemental Table S8). Only one co-occurrence between genes was filtered out. Network visualization and hierarchical community clustering was conducted using the linkcomm package (Kalinka and Tomancak 2011) in the R software (R Core Team 2018). The network was weighted by the number of co-occurrence between strains.
Analyses of genome sizes, prophages, and integrases
Genome sizes were estimated from the total scaffold length of each strain. Prophages and integrases were detected in each draft genome sequence using VirSorter (Roux et al. 2015) and PhiSpy (Akhter et al. 2012), respectively.
Identification of lineage-associated genes
The pan-genome matrix generated from the pan-genome analysis using Roary with options (-i 80 -cd 100 -s) was used as an input for pan-GWAS analysis by Scoary (Brynildsrud et al. 2016) to identify genes associated with either of the bovine- or human-associated lineages. Statistical significance was corrected for multiple comparisons with the Bonferroni method. The same analyses were performed to compare gene repertoires between phylogroups B1 and B2 strains and identify genes associated with either of B1 or B2 strains.
Statistical analyses
All statistical analyses were performed using R version 3.3.2 (R Core Team 2018). To assess the coexistence of each virulence gene with LEE or stx and the difference of effector conservation between LEE-positive human clinical isolates and LEE-positive bovine commensal isolates, statistical significance was determined by the Fisher's exact test with the Bonferroni correction for multiple comparisons. Statistical significance of the differences in genome sizes and numbers of phages and integrases between the LEE/stx-, stx-, LEE-positive strains and LEE/stx-negative strains was assessed based on a generalized linear model (GLM) with a negative binomial distribution and log-link function (glm.nb in the library MASS in R).
Data access
All sequence data generated in this study have been submitted to the NCBI BioProject database (BioProject; https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJDB5579.
Acknowledgments
This work was funded by JSPS KAKENHI under grant numbers 16H06279, 16K15278, and 17H04077 to Y.O. and 16H05190 to T.H. and by AMED under grant numbers JP17efk0108127h0002 to Y.O. and 18fk0108065h0801 to T.H. We thank A. Yoshida, H. Iguchi, Y. Inoue, M. Shimbara, M. Horiguchi, and Y. Sato for providing technical assistance. We also thank H. Funakura, Y. Inoue, K. Toda, S. Terayama, Z. Nakamura, H. Hasunuma, D. Matsumoto, Y. Iwatani, A. Kunisawa, and I. Kobayashi for collecting cattle stool samples.
Author contributions: Y.O. and T.H. designed the study. Y.O., Y.K., K.U., T.S., S.Y., T.O, A.I., T.M.-I., M.O., F.A., H.B., E.O., J.G.M., K.S.A., and D.D. collected the samples. Y.A., Y.O., M.P.S., Y.G., Y.T., and Y.N. analyzed the data. Y.A., T.H., and Y.O. wrote the manuscript. Y.O., K.A., and T.H. were responsible for supervision and management of the study.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.249268.119.
-
Freely available online through the Genome Research Open Access option.
- Received February 12, 2019.
- Accepted July 3, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








