
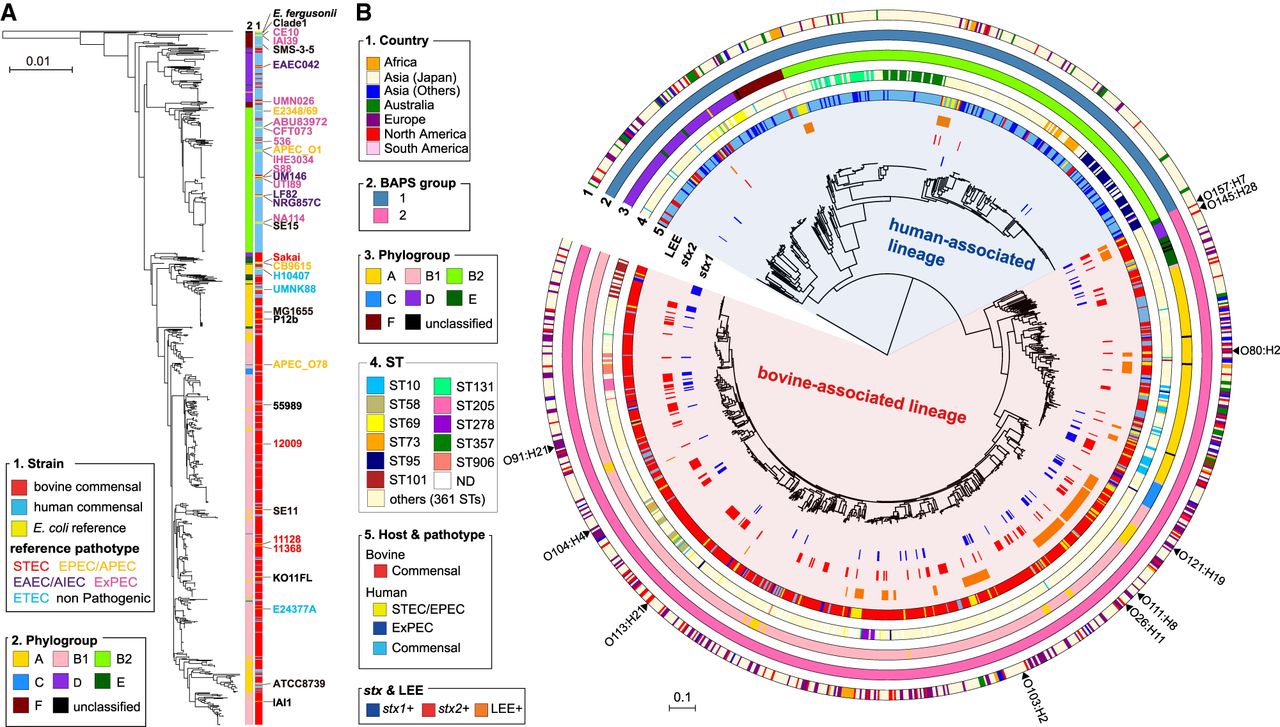
Phylogenetic relationship between human and bovine commensal E. coli and distribution of stx- and LEE-positive isolates. (A) The NJ tree of 937 bovine and human commensal isolates with 36 completely sequenced reference strains based on the concatenated nucleotide sequences of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA). The phylogeny was rooted using Escherichia fergusonii strain ATCC35469 and cryptic Escherichia clade 1 strain TW15838. Sources of sequences and phylogroups of strains are shown in the NJ tree. Pathotypes of reference strains are indicated by differently colored characters. (B) The core gene–based ML tree of 937 bovine and human commensal isolates with 197 human clinical isolates. Cryptic Escherichia clade I strain TW15838 was included as an outgroup. The tree was constructed based on 247,627 SNPs located on 1755 core genes. Country, BAPS group, phylogroup, ST, host and pathotype of each strain, and the presence of stx1, stx2, and LEE are shown. Positions of clinical isolates of major STEC serotypes are indicated.








