
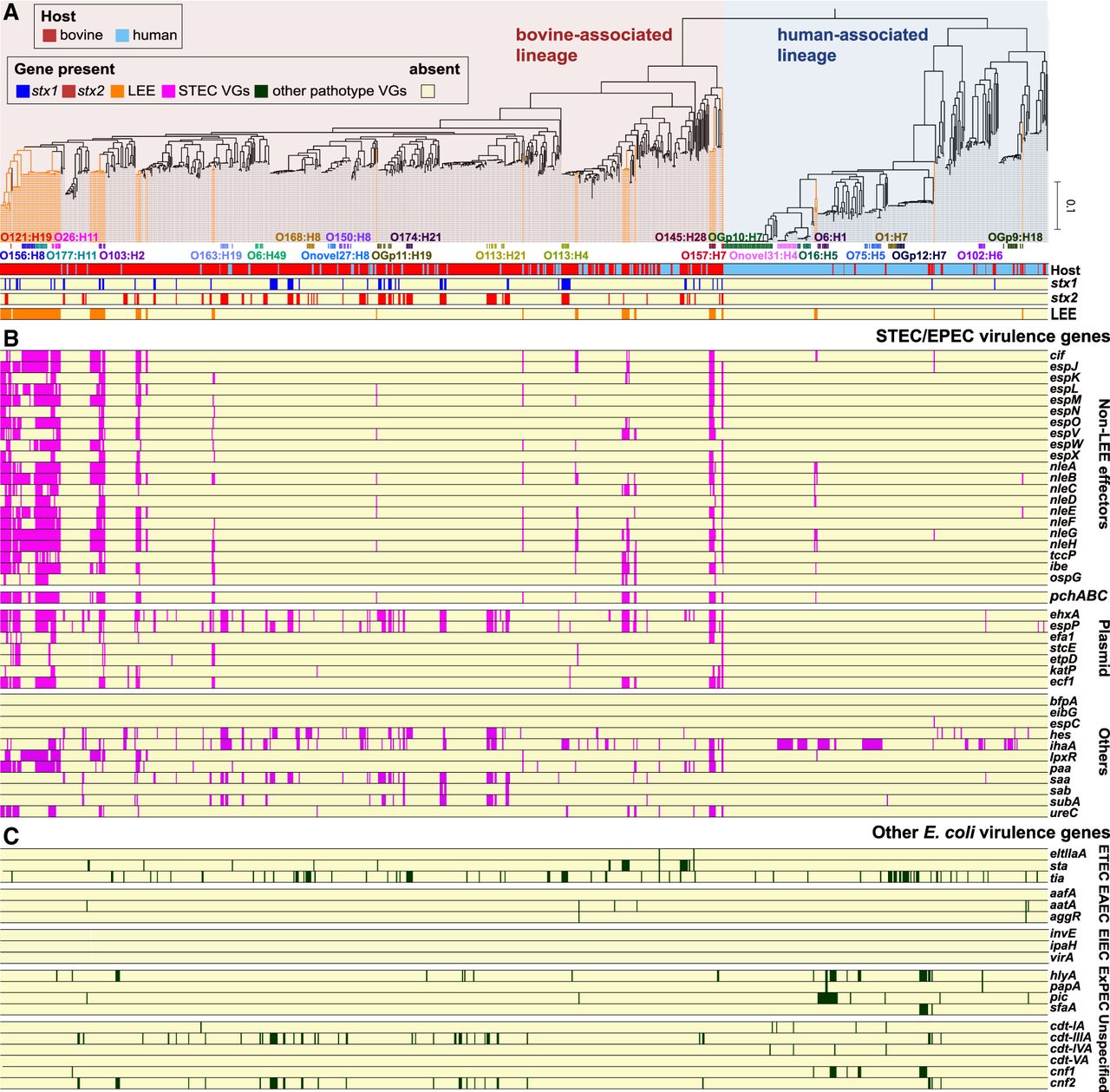
Figure 2.
Distribution of virulence genes among 937 bovine and human commensal E. coli strains. (A) The ML tree based on 262,788 SNP sites on 1958 core genes. The tree was rooted by cryptic Escherichia clade I strain TW15838. LEE-positive lineages/strains are highlighted by orange lines. Frequently observed serotypes (more than four strains) and major STEC serotypes are indicated. The presence of stx1, stx2, and LEE in each strain is shown. The presence of other STEC/EPEC virulence genes (B) and virulence genes associated with other E. coli pathotypes (C) is shown, respectively. For the functions and nucleotide sequences of each gene analyzed, see Supplemental Table S7.








