
Estrogen-independent molecular actions of mutant estrogen receptor 1 in endometrial cancer
Abstract
Estrogen receptor 1 (ESR1) mutations have been identified in hormone therapy–resistant breast cancer and primary endometrial cancer. Analyses in breast cancer suggest that mutant ESR1 exhibits estrogen-independent activity. In endometrial cancer, ESR1 mutations are associated with worse outcomes and less obesity, however, experimental investigation of these mutations has not been performed. Using a unique CRISPR/Cas9 strategy, we introduced the D538G mutation, a common endometrial cancer mutation that alters the ligand binding domain of ESR1, while epitope tagging the endogenous locus. We discovered estrogen-independent mutant ESR1 genomic binding that is significantly altered from wild-type ESR1. The D538G mutation impacted expression, including a large set of nonestrogen-regulated genes, and chromatin accessibility, with most affected loci bound by mutant ESR1. Mutant ESR1 is distinct from constitutive ESR1 activity because mutant-specific changes are not recapitulated with prolonged estrogen exposure. Overall, the D538G mutant ESR1 confers estrogen-independent activity while causing additional regulatory changes in endometrial cancer cells that are distinct from breast cancer cells.
Estrogen receptor 1 (ESR1) is a ligand-inducible steroid hormone receptor that acts as an oncogene in many breast and endometrial tumors. In these diseases, hormone therapies can be used to reduce estrogen signaling either through a reduction in estrogen production or a reduction in ESR1 activity. Mutations in the ligand binding domain (LBD) of ESR1 have been associated with hormone therapy resistance in breast cancer (Fuqua et al. 1993; Osborne and Schiff 2011; Robinson et al. 2013; Toy et al. 2013, 2017; Fuqua et al. 2014; Jeselsohn et al. 2014) and a recent large-scale analysis of ESR1 mutations found that 14% of metastatic breast cancers harbor a LBD mutation (Toy et al. 2017). ESR1 LBD mutations were not identified in primary tumors in The Cancer Genome Atlas’ study on breast cancer (The Cancer Genome Atlas Network 2012), indicating that ESR1 mutations are not observed at clonal frequencies and are unlikely to play a role in tumor initiation; however, ESR1 LBD mutations can be found at low mutation frequencies in primary breast tumors (Toy et al. 2017). In contrast, heterozygous ESR1 LBD mutations are found in 5.8% of primary endometrial cancers with endometrioid histology (The Cancer Genome Atlas Research Network 2013; Backes et al. 2016; Gibson et al. 2016), representing approximately 3500 new uterine cancer diagnoses with an ESR1 LBD mutation in the United States each year. The presence of ESR1 mutations is associated with obesity-independent endometrial cancer, and patients with ESR1 LBD mutations trend toward worse prognosis when compared to patients with wild-type ESR1 tumors (Backes et al. 2016).
The ESR1 LBD mutations occur in a region of the protein essential for ligand binding and interactions with coregulatory proteins, with the majority of mutations found at residues D538 and Y537. Studies into the molecular and phenotypic consequences of ESR1 LBD mutations have been performed in breast cancer, revealing that the mutations confer estrogen-independent ESR1 activity, which drives gene regulation and cell proliferation in the absence of estrogens (Merenbakh-Lamin et al. 2013; Robinson et al. 2013; Toy et al. 2013, 2017; Jeselsohn et al. 2014, 2018; Bahreini et al. 2017; Zhao et al. 2017). Biochemical characterization of the mutations suggests that mutant ESR1 favors the activated conformation of the receptor irrespective of ligand, causing constitutive receptor activity (Merenbakh-Lamin et al. 2013; Fanning et al. 2016; Toy et al. 2017; Zhao et al. 2017; Katzenellenbogen et al. 2018). Gene expression analyses highlight the ability of mutant ESR1 to regulate canonical ESR1 target genes in the absence of estrogens (Merenbakh-Lamin et al. 2013; Robinson et al. 2013; Toy et al. 2013, 2017; Jeselsohn et al. 2014, 2018; Bahreini et al. 2017; Katzenellenbogen et al. 2018). In addition to ligand-independent regulation of genes that are normally impacted by 17β-estradiol (E2), novel non-E2-regulated genes are also affected by mutant ESR1 (Bahreini et al. 2017; Jeselsohn et al. 2018), suggesting that these mutations may confer additional functionality to ESR1 than just constitutive activity. Although these studies have uncovered important features of the molecular and phenotypic consequences of ESR1 mutations in breast cancer, similar analyses have not been performed in endometrial cancer cells. Because gene expression responses to estrogens and ESR1 genomic binding are highly dissimilar between breast and endometrial cancer (Gertz et al. 2012, 2013; Droog et al. 2017), the impact of ESR1 LBD mutations in endometrial cancer cells could be different than the effects observed in breast cancer cells.
In this study, we sought to gain an understanding of the molecular consequences of ESR1 LBD mutations in endometrial cancer. Using Ishikawa cells, a human endometrial adenocarcinoma cell line that is a cell culture model for type I disease, we used a CRISPR/Cas9-mediated epitope tagging strategy. We created endometrial cancer cells that are heterozygous for the D538G ESR1 ligand binding domain mutation (or wild-type ESR1 for controls) coupled with a FLAG epitope tag incorporated at the C terminus of the endogenous locus. The addition of an epitope tag to the endogenous gene allowed us to specifically analyze binding of the mutant form of ESR1 by chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq). We explored mutation-specific gene expression effects via RNA-seq, assessed changes to the chromatin landscape using the assay for transposase accessible chromatin followed by sequencing (ATAC-seq), and analyzed effects on proliferation and migration. We also investigated whether the regulatory effects of mutant ESR1 could be recapitulated by prolonged exposure to E2. The systematic investigation of mutant ESR1's molecular activity in endometrial cancer cells will enable future phenotypic and mechanistic investigation into ESR1 mutant endometrial cancer.
Results
Generation of D538G ESR1 mutant and wild-type cell lines
The molecular consequences of ESR1 LBD mutations have not been explored in endometrial cancer and warrant investigation. We used a CRISPR/Cas9-mediated epitope tagging strategy called CETCH-seq (Savic et al. 2015) in Ishikawa cells, an endometrial adenocarcinoma cell line that exhibits ESR1 genomic binding similar to endometrial tumor samples (Rodriguez et al. 2019a), to model a common ESR1 LBD mutation, D538G. This technique combines guide RNAs that target Cas9 to the C terminus of ESR1 and a donor plasmid that leads to the incorporation of a 3X FLAG epitope tag and neomycin resistance gene at ESR1’s endogenous locus (Fig. 1A). Ishikawa cells were first transfected with plasmids, treated with G418 to select for resistant cells and subjected to limiting dilution plating to generate single-cell clones. Using this technique, we generated three cell lines that were heterozygous for the D538G ESR1 mutation on a FLAG-tagged allele and two heterozygous FLAG-tagged wild-type cell lines (referred to as wild-type clones throughout; the original Ishikawa cell line is referred to as parental). FLAG and ESR1 protein expression were established via western blot (Fig. 1B), and D538G mutations were confirmed by Sanger sequencing. Additionally, we observed similar expression frequencies of the wild-type and mutant alleles in the D538G clonal cell lines by RNA sequencing (RNA-seq) (Fig. 1C). The creation of these isogenic cell lines enabled further studies into the gene regulatory changes caused by the D538G ESR1 mutation.

Generation and characterization of ESR1 wild-type and D538G mutant models. (A) CRISPR-mediated epitope tagging strategy was used to generate heterozygous FLAG-tagged wild-type ESR1 and D538G mutant ESR1 Ishikawa cell lines. (B) Immunoblotting for FLAG and ESR1 in Ishikawa parental cells, two heterozygous FLAG-tagged wild-type and three heterozygous FLAG-tagged D538G mutant cell lines show protein expression of epitope-tagged ESR1 and total ESR1. (C) The ESR1 wild-type and mutant allele expression frequencies based on RNA-seq data is shown for each D538G clonal cell line. (D) Estrogen response element (ERE) reporter activity as measured by luciferase activity was assayed in DMSO- and E2-induced conditions. Experiments were performed in triplicate, and the average luciferase activity for two wild-type and three D538G mutant clones is shown. (***) P = 0.0002; (****) P < 0.0001; error bars represent SEM.
D538G mutant ESR1 displays ligand-independent regulatory activity
To assess ESR1's transcriptional activity in our models, we cultured wild-type and D538G mutant cells in hormone-deprived media for 5 d and then transfected the cell lines with a luciferase estrogen response element (ERE) reporter assay. One day post-transfection, cells were treated with either vehicle (DMSO) or 10 nM E2 for 24 h before measuring luciferase activity. We observed negligible luciferase expression in the wild-type cells treated with DMSO (Fig. 1D). However, there was a significant 30-fold increase in expression in wild-type cells induced with E2. We detected ESR1 activity in the D538G mutant clones treated with DMSO at 30% of the wild-type E2 levels. Activity was significantly increased in the D538G mutant lines treated with E2, with levels similar to the wild-type lines treated with E2. These results indicate ligand-independent transcriptional activity of the D538G mutant in endometrial cancer cell lines.
Introduction of the D538G ESR1 mutation causes large transcriptional changes
The ligand-independent transcriptional activity of mutant ESR1 in the reporter assay suggested that transcription in these mutant lines could be altered, leading to aberrant gene expression. To uncover gene expression changes caused by the mutation, we performed RNA-seq on the wild-type FLAG-tagged clones and D538G mutant clones following an 8-h 10 nM E2 induction in hormone-deprived media. Principal component analysis of the isogenic lines clustered wild-type and mutant lines separately, with the first principal component accounting for 46% of the variance in our data sets while separating samples based on the presence of the mutation (Supplemental Fig. S1A). Treatment with E2 along with differences between clones with the same genotype accounted for 29% of the variance in these lines and is represented in the second principal component, highlighting the importance of analyzing multiple clones to capture the variance in clone derivation. Our analysis indicates that the gene expression profile of wild-type cells supplemented with E2 did not recapitulate the expression changes seen in the D538G mutant lines.
Analysis of differentially expressed genes across clonal cell lines revealed multiple expression patterns (Fig. 2A; for gene lists, see Supplemental Table S1). We identified 119 genes that were up-regulated and 48 genes that were down-regulated in response to an estrogen induction in wild-type cells (adjusted P-value <0.05). A comparison of these E2 responsive genes to genes regulated in the D538G DMSO-treated lines revealed 47 up-regulated and 10 down-regulated genes that were responsive to estrogen in wild-type cells and also regulated by the D538G mutation independent of E2. Examples of mutant ESR1 ligand-independent regulation of estrogen-responsive genes confirmed by qPCR include progesterone receptor (PGR) known to play critical roles in reproductive function (Fig. 2B) and a matrix metallopeptidase (MMP17), which has been implicated in the degradation of the extracellular matrix (Fig. 2C). The estrogen-independent regulation of these genes by mutant ESR1 is consistent with the reporter assays and the hypothesis that mutant ESR1 has ligand-independent activity.
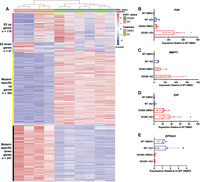
D538G mutant drives a distinct transcriptional program. (A) Heatmap shows the relative expression of E2 up- and down-regulated genes as well as mutant-specific differentially expressed genes. Sample types are indicated by the column annotations described in the legend. Validation of RNA-seq results by qPCR are shown for ligand-independent E2–up-regulated genes PGR (B) and MMP17 (C) as well as mutant-specific up-regulated gene EHF (D) and down-regulated gene EPHA3 (E). B–E show average expression levels, normalized to wild-type cells without E2 treatment, for two ESR1 wild-type and three D538G mutant clones after 8-h E2 or DMSO (vehicle) induction. Error bars represent SEM.
In addition to changes in estrogen-regulated genes, the D538G mutation impacted the expression of many genes not normally regulated by estrogen. We identified mutation-specific gene expression changes with 302 novel genes that are up-regulated and 241 novel genes that are down-regulated owing to the mutation (for mutant-specific genes, see Supplemental Table S1). Examples of novel genes include EHF, an ETS factor not normally expressed in Ishikawa cells (Fig. 2D) and EPHA3, a receptor tyrosine kinase (Fig. 2E). Ingenuity pathway analysis indicated that novel genes were enriched for pathways associated with more aggressive tumors, which include cellular growth, proliferation, and movement (Supplemental Fig. S1B,C). This pathway enrichment is similar to findings from studies performed on ESR1 mutations in MCF-7 and T47D, two breast cancer cell lines. Although the pathways are consistent with novel gene regulation observed in breast cancer cells, there is little overlap in the differentially expressed genes from Bahreini et al. (2017) (MCF-7: 29 genes [5.3%]; T47D 20 genes [3.7%]) and Jeselsohn et al. (2018) (MCF-7: 91 genes [16.7%]). Collectively, our data indicate that the D538G mutation impacts many non-E2 regulated genes and enables a more expansive and potentially aggressive transcription program.
Although there are not enough ESR1 mutant patient samples with associated gene expression data available to determine whether the observed mutant-specific gene expression changes are seen in endometrial tumors, we can determine if the expression of these mutant-specific genes are associated with patient outcomes. To explore this connection, we analyzed gene expression and disease-free survival in the Cancer Genome Atlas (TCGA) endometrial cancer cohort (The Cancer Genome Atlas Research Network 2013) and restricted the analysis to high ESR1-expressing tumors with endometrioid histology. We found significant overlaps between mutant regulated genes and genes whose expression is associated with disease-free survival (Supplemental Fig. S2, examples; Supplemental Table S2, full gene list). For the mutant up-regulated genes, there was 3.2-fold enrichment specifically for genes associated with worse outcomes (P-value = 2.7 × 10−4, Fisher's exact test) (Supplemental Fig. S2). The mutant down-regulated genes were enriched 2.8-fold over random chance specifically in genes whose expression was associated with longer disease-free survival (P-value = 6.9 × 10−3, Fisher's exact test) (Supplemental Fig. S2). These results suggest that genes regulated by mutant ESR1, that are unrelated to E2 inductions, tend to be associated with outcomes in endometrial cancer patients in a pattern consistent with mutant ESR1 driving more aggressive tumors.
D538G mutation increases migration without altering proliferation
The cellular and molecular pathways found enriched in D538G mutant-specific gene lists suggested a more aggressive phenotype in endometrial cancer cells. To determine how the mutation affected proliferation, wild-type and D538G mutant cells were seeded at low densities in full serum media and hormone-deprived media for up to 72 h. Growth rates were measured on the IncuCyte ZOOM live-cell imaging platform. The D538G mutation did not enhance proliferation in full serum media when compared to ESR1 wild-type cells (Fig. 3A), with all cell lines showing similar growth rates. In hormone-deprived media, although we observed differences in growth rates between clones, there were not consistent differences between ESR1 wild-type and D538G mutant lines. We also tested the D538G mutant's ability to affect migration via a wound healing assay. ESR1 wild-type and D538G mutant cells were initially grown to 100% confluency in full serum and hormone-deprived media, the cell monolayer was then scraped and imaged for up to 24 h on the IncuCyte ZOOM. The D538G mutation enhanced migration by 40.3% compared to wild-type lines in full serum media (P-value <0.0001, unpaired t-test) (Fig. 3B,C) and by 35.4% in hormone-deprived media (P-value <0.0001, unpaired t-test) (Fig. 3D,E), suggesting that the mutation confers a more migratory phenotype. These results are consistent with mutant ESR1 contributing to more aggressive endometrial tumors.
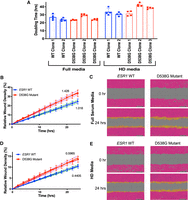
D538G mutant affects migration but not proliferation in endometrial cancer cells. (A) Bar graphs indicating the doubling times for ESR1 wild-type and D538G mutant cell lines in full media and hormone-deprived (HD) media. The relative wound densities of two ESR1 wild-type and three D538G mutant cell lines are shown over 24 h following scratch/wounding of cell monolayer in full serum media (B) and HD media (D). Images show the initial wound (pink) and migratory cells (orange) in wild-type and D538G mutant cells at 0 and 24 h in full serum media (C) and HD media (E). Proliferation and migration figures represent at least three independent experiments, performed in triplicate. Error bars represent SEM.
D538G mutation alters ESR1 genomic binding
To ascertain ESR1 genomic binding sites in Ishikawa cells and the manner in which the D538G mutation alters ESR1's genomic interactions, we performed ChIP-seq with an antibody that recognizes the FLAG epitope tag in the two wild-type and three mutant clones after 1 h treatments with DMSO or E2. We identified <55 peaks in each of the two wild-type lines treated with DMSO, indicating negligible ESR1 binding in the absence of E2 treatment. We observed a significant increase in genomic binding, with 20,104 ESR1 binding sites (ERBS) on average identified in the two wild-type cell lines following E2 induction. An analysis of the overlap of binding sites called in the wild-type clones treated with E2 showed 90% (15,780 out of 17,466 on average across replicates) concordance between lines, highlighting the reproducibility of our experimental findings (Supplemental Fig. S3A). To confirm that the FLAG tag did not significantly alter ESR1 genomic binding, we overlapped the binding sites discovered with the FLAG antibody to binding sites from a ChIP-seq previously performed in Ishikawa cells with an antibody that recognizes ESR1 (Gertz et al. 2012, 2013). We observed 77% overlap (5583 out of 7294) on average with this previously generated ESR1 ChIP-seq data set, indicating that the epitope tag does not drastically affect ESR1 binding in the wild-type cell lines (Supplemental Fig. S3B–E). In contrast, we observed D538G mutant ESR1 binding at >22,292 binding sites on average in the three mutant lines in the absence of E2, suggesting that the mutation confers ligand-independent binding in endometrial cancer cell lines.
Analysis of differential binding between wild-type clones treated with E2 and all D538G mutant clone samples established 6534 constant ESR1 binding sites, 2205 binding sites that were enriched in the D538G mutant lines, and 3316 sites that were enriched in the wild-type lines (Fig. 4A; Supplemental Fig. S4). When comparing loci enriched in D538G ESR1 binding to loci enriched in wild-type ESR1 binding, we found a 1.30-fold enrichment in intronic regions (P-value = 3.2 × 10−6, Fisher's exact test), a 1.96-fold depletion in promoter regions (P-value = 1.3 × 10−8, Fisher's exact test), and a 1.30-fold depletion in intergenic regions (P-value = 4.2 × 10−6, Fisher's exact test) (Supplemental Fig. S3F). Using the loci enriched in D538G ESR1 binding, de novo motif analysis revealed a significant enrichment for the canonical ERE (P-value = 8.5 × 10−18, MEME) (Bailey et al. 2009), indicating that many of these novel sites are direct targets of the D538G mutant (Supplemental Fig. S3G). Additional motifs were also enriched at these sites including the Zic family of transcription factors (P-value = 5.7 × 10−16, MEME), the NF-I transcription factors (P-value = 2.5 × 10−17, MEME) (Bailey et al. 2009), and E-box–binding proteins (P-value = 1.7 × 10−11, MEME) (Supplemental Fig. S3G). The sites enriched in ESR1 binding in the wild-type lines were also enriched in the canonical ERE motif (P-value = 1.9 × 10−47, MEME), although other motifs were enriched, including the forkhead factor motif (P-value = 4.1 × 10−35, MEME) and the Sox factor motif (P-value = 6.3 × 10−27, MEME) (Supplemental Fig. S3H). Although both mutant-enriched and wild-type–enriched ESR1 bound loci had enrichment of EREs, we found that the strength of EREs found in constant ESR1 binding sites and wild-type–enriched ESR1 binding sites was significantly higher than EREs found in D538G enriched sites (P-value <2.2 × 10−16, Wilcoxon signed-rank test) (Fig. 4B). These results indicate that the D538G mutation alters ESR1 genomic binding, including a move to sites with suboptimal EREs.
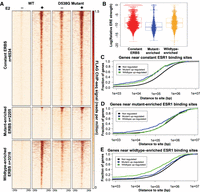
D538G mutation alters ESR1 genomic binding. (A) Heatmap displays ESR1 binding in representative wild-type and D538G mutant clones, in which each row is an ESR1 binding site. The heatmaps include sites that are constant in wild-type and mutant lines (top), sites that are enriched in the mutant lines (middle), and sites that are enriched in wild-type lines (bottom). (B) Plot shows that the distribution of the predicted relative affinity for ESR1, based on the best match to an ERE, is higher in constant binding sites (red) and wild-type–enriched sites (yellow) than D538G mutant-enriched ESR1 binding sites (blue). Cumulative distribution plots show the fraction of mutant up-regulated, down-regulated, or not regulated genes that have a constant (C), mutant-enriched (D), or wild-type–enriched (E) ESR1 binding site within a given distance from the transcription start site.
The regulatory proteins used by ESR1 to mediate gene expression in breast cancer cells have been well characterized by multiple laboratories (Carroll et al. 2005; Hurtado et al. 2011; Magnani et al. 2011; Tan et al. 2011; Mohammed et al. 2013). However, the proteins responsible for mediating these interactions in endometrial cancer have not been established, but ETV4 (Gertz et al. 2013) and FOXA1 (Droog et al. 2017) have been proposed as key transcription factors. To understand the relationship between mutant ESR1 binding and these factors, we overlapped ESR1 binding sites with the binding sites of FOXA1, which is reported to overlap with 8% of ESR1's binding sites in parental Ishikawa cells, and ETV4, which is reported to overlap with 45% of ESR1's binding sites in parental Ishikawa cells (Gertz et al. 2013). We found that mutant-enriched ESR1 binding sites are depleted in FOXA1 binding sites (P-value = 1.0 × 10−4, Fisher's exact test) compared to wild-type–enriched ESR1 bound sites. In addition, mutant-enriched binding sites were also significantly depleted in ETV4's binding sites (P-value <2.2 × 10−16, Fisher's exact test) compared to wild-type-enriched ESR1 bound sites, suggesting that other transcription factors might be playing a role in mutant ESR1 genomic binding.
To explore the connection between mutant-enriched ESR1 binding and gene expression, we analyzed the distance between mutant-enriched, constant, and wild-type–enriched ESR1 binding sites and the transcription start sites (TSS) of genes up-regulated, down-regulated, or not regulated by the mutation. We found that constant binding sites were closer to both mutant up-regulated (P-value = 1.51 × 10−15, Wilcoxon signed-rank test) and mutant down-regulated genes (P-value = 6.2 × 10−14, Wilcoxon signed-rank test), than not regulated genes (Fig. 4D). Mutant-enriched binding sites were significantly closer to mutant up-regulated genes (P-value = 7.7 × 10−15, Wilcoxon signed-rank test) (Fig. 4C) and wild-type–enriched sites were found closer to genes that were down-regulated by the mutation (P-value = 6.2 × 10−16, Wilcoxon signed-rank test) (Fig. 4E). Overall, the association between genes and ESR1 binding sites suggests that wild-type and mutant ESR1 are generally acting as activators and may be directly contributing to many of the mutant-specific gene expression changes. The association of constant ESR1 binding sites with both mutant up-regulated and down-regulated genes implies that these ESR1 bound sites may be becoming more or less active when bound by mutant ESR1 compared to wild-type ESR1.
D538G ESR1 mutation leads to changes in accessible chromatin
The mutant-specific gene expression changes and ESR1 binding alterations led us to hypothesize that the D538G mutation may affect chromatin accessibility. To test this hypothesis, we performed the assay for transposase accessible chromatin followed by sequencing (ATAC-seq) in the wild-type and D538G mutant cell lines, after a 1-h treatment with DMSO or E2. Similar to the distinct RNA-seq profiles, principal component analysis of the isogenic lines once again clustered wild-type and mutant lines separately (Fig. 5A). The first principal component accounted for 60% of the variance in the ATAC-seq data and separated wild-type and D538G mutant lines. The second principal component, which accounted for 18% of the variance, separated clones, but E2 treatment for 1 h was not a large contributor to the variance. This analysis suggests that the 1-h E2 induction does not have major genome-wide effects on chromatin accessibility in comparison to the D538G ESR1 mutation.
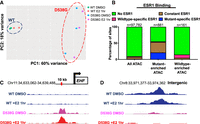
ESR1 D538G mutation alters chromatin accessibility at multiple loci. (A) Principal component analysis shows the relationship between ATAC-seq signal of ESR1 wild-type (blue circle) and D538G mutant cell lines (red circle). (B) Less than 10% of all ATAC-seq sites overlap with ESR1 binding sites, whereas 55% of mutant-enriched ATAC-seq sites overlap ESR1 binding sites, including constant ESR1 binding (brown) and mutant-enriched ESR1 binding (blue). Fifteen percent of wild-type–enriched ATAC-seq sites overlap ESR1 binding sites. Representative browser tracks show ATAC-seq signal increases with the D538G mutation at a region near EHF (C) and ATAC-seq signal decreases at an intergenic region on Chromosome 8 (D). Wild-type ATAC-seq signal DMSO/+E2 (blue), D538G DMSO (pink), and D538G + E2 (red) are scaled to the same value at each locus.
Analysis of ATAC-seq signal from our experiments identified 881 regions that were significantly more accessible and 161 regions that were significantly less accessible in the D538G mutant cell lines compared to wild-type cell lines. Wild-type–enriched regions were 14.3-fold more likely to reside in promoter regions compared to mutant-enriched regions (P-value <2.2 × 10−16, Fisher's exact test), whereas mutant-enriched regions were 1.6-fold more likely to reside in intronic regions (P-value = 0.0098, Fisher's exact test) (Supplemental Fig. S7B). Examples of these mutant-enriched and mutant-depleted ATAC-seq regions are shown in Figure 5, C and D, and a heatmap of the mutant-enriched sites shows large magnitude changes in ATAC-seq signal at the vast majority of these loci (Supplemental Fig. S5A). We found that although 9.6% of all identified open chromatin regions are associated with ESR1 binding, ESR1 binding was found at the majority of mutant-enriched ATAC-seq sites, with 55% of the mutant-enriched ATAC-seq sites exhibiting ESR1 binding, including constant and mutant-enriched ESR1 binding sites (Fig. 5B). Motif analysis at ESR1-associated, mutant-enriched ATAC-seq sites identified the ERE motif as expected (P-value = 3.38 × 10−23, MEME) and the NF-I motif (P-value = 6.64 × 10−27, MEME) (Supplemental Fig. S5B). These results suggest that mutant ESR1 could be playing a major role in chromatin accessibility, possibly owing to mutant ESR1's constitutive activity. We found that 45% of mutant-enriched ATAC-seq sites were not associated with ESR1 binding and these sites were enriched for TEAD4 (P-value = 6.41 × 10−10, MEME) and AP-1 (P-value = 4.28 × 10−9, MEME) motifs (Supplemental Fig. S5C), suggesting that other transcription factors may be contributing to mutant-specific alterations in chromatin accessibility.
Prolonged exposure to estrogen does not re-create mutant-specific regulatory effects
The mechanism by which mutant ESR1 regulates a novel set of genes may be explained by constitutive ESR1 activity or neomorphic functions conferred by the D538G mutation. To determine how much of the gene regulatory effects of mutant ESR1 are attributable to constitutive ESR1 activity, we cultured the two wild-type clones in the presence of 10 nM E2 for a 25-d period and performed RNA-seq and ATAC-seq on cells collected at 10-, 15-, 20-, and 25-d intervals during this prolonged exposure. Differential gene expression analysis identified 658 genes that were up-regulated and 1138 genes that were down-regulated in prolonged E2-treated wild-type cells compared to wild-type cells treated for 8 h with E2 or DMSO (Fig. 6A). These numbers suggest there are more genes regulated by prolonged exposure to E2 than the transient 8-h induction; however, the overlap with mutant-specific gene expression changes was minimal. Only 25 genes (8.3% of mutant-specific up-regulated genes) that were up-regulated in response to prolonged E2 were also up-regulated by the mutation (Fig. 6B; Supplemental Fig. S6, examples). Additionally, only 34 down-regulated genes (14% of mutant-specific down-regulated genes) overlapped with mutant-specific down-regulated genes (Fig. 6C). These findings suggest that the D538G mutant regulates a ligand-independent transcriptional program that is dissimilar to prolonged E2 exposure in endometrial cancer cells.
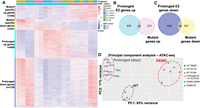
Prolonged E2 exposure does not recapitulate D538G mutant regulatory consequences. (A) Heatmap shows the relative expression of mutant-specific differentially expressed genes as well as genes up- and down-regulated in response to prolonged E2 (each row is a gene). Samples are indicated by the column annotations described in the legend. (B) Venn diagram shows the overlap between genes up-regulated in wild-type lines exposed to prolonged E2 and mutant-specific up-regulated genes. (C) Venn diagram shows the overlap between genes down-regulated in wild-type lines exposed to prolonged E2 and mutant-specific down-regulated genes. (D) Principal component analysis of ATAC-seq signal exhibits three sample groups: wild-type lines with 1-h or no E2 treatment (navy circle), wild-type lines with prolonged E2 exposure (gray circle, numbers indicate days of treatment), and D538G mutant lines (red circle).
To determine if constitutive ESR1 activity could re-create the mutant-specific chromatin accessibility patterns, we compared the ATAC-seq results from the prolonged E2 experiment in wild-type cells to 1- and 8-h E2 inductions in wild-type cells as well as the results from the D538G mutant clones. Principal component analysis of all ATAC-seq samples identified three distinct clusters: (1) DMSO controls along with short-term E2-treated wild-type cells (1 h); (2) prolonged E2-treated wild-type cells (10, 15, 20, and 25 d) along with the 8-h E2 induction; and (3) the D538G mutant lines (Fig. 6D). Although Figure 5A indicates that the 1-h E2 induction did not significantly affect chromatin accessibility globally, prolonged E2 exposure is able to affect chromatin accessibility, with some features being shared with the mutation. However, because the prolonged E2-treated cells do not cluster with the mutant lines, we can conclude that these features do not recapitulate most of the mutant-specific genome-wide effects on chromatin accessibility.
The ATAC-seq analysis revealed 1488 regions that became more accessible and 2915 regions that were less accessible following prolonged E2 exposure in wild-type cells (Supplemental Fig. S7). Regions more accessible upon prolonged E2 treatment were 20.8-fold more likely to reside in promoter regions compared to regions less accessible after prolonged E2 treatment (P-value <2.2 × 10−16, Fisher's exact test), whereas less accessible regions were 3.7-fold more likely to reside in intergenic regions (P-value <2.2 × 10−16, Fisher's exact test) and 2.5-fold more likely to reside in intronic regions (P-value <2.2 × 10−16, Fisher's exact test) (Supplemental Fig. S7C). We overlapped regions that are more accessible after prolonged E2 exposure and regions that are more accessible in the D538G mutant clones, as described above, and identified only 77 regions that were common between data sets, which represented 8.7% of the mutant-enriched regions. Similarly, there were 44 loci that overlapped between chromatin that becomes less accessible in response to prolonged E2 treatment and mutant-depleted accessible chromatin, representing 27% of the mutant-depleted regions (Supplemental Fig. S6, examples). Our findings indicate that prolonged E2 is unable to recapitulate the observed ESR1 mutant-driven changes to chromatin accessibility. Collectively, the effects on gene expression and chromatin accessibility both suggest that the D538G mutation confers distinct neomorphic properties to the receptor that cannot be adequately explained by constitutive estrogen signaling.
Discussion
Through the creation of isogenic models of a common ESR1 mutation, D538G, we found that mutant ESR1 exhibits estrogen-independent activity in endometrial cancer cells. Similar to findings in breast cancer (Merenbakh-Lamin et al. 2013; Robinson et al. 2013; Toy et al. 2013; Jeselsohn et al. 2014, 2018; Bahreini et al. 2017), mutant ESR1 is able to bind the genome and drive the transcription of estrogen-responsive genes in the absence of estrogens. The estrogen-independent activity of mutant ESR1 that we observed is consistent with the clinical observation that endometrial cancer patients with ESR1 mutations have lower body mass index than patients without ESR1 mutations (Backes et al. 2016). Adipose tissue, which is more prevalent in obese patients, is capable of peripheral estrogen production (Siiteri 1987), but endometrial tumors with ESR1 mutations appear not to rely on this excess estrogen, presumably owing to constant ESR1 activity (Rodriguez et al. 2019b).
The use of multiple ESR1 mutant and wild-type clones enabled the discovery of molecular changes that can be reproducibly attributed to the mutation. Wild-type ESR1 binds to different loci in breast cancer and endometrial cancer cells (Gertz et al. 2013) and primary tumors (Droog et al. 2017) leading to different transcriptional responses to E2. Mutant ESR1 exhibits a similar cell-type–specific pattern in which the genes regulated by mutant ESR1 are different between endometrial cancer cells and breast cancer cells. This is true for both estrogen-independent regulation of normally estrogen-responsive genes as well as novel regulation by mutant ESR1 of nonestrogen-responsive genes. These results suggest that mutant ESR1 binding site and target gene selection is still constrained by the other transcription factors and cofactors expressed in the cell as well as the chromatin landscape. Although different genes are regulated by mutant ESR1 in breast and endometrial cancer cells, similar pathways including cellular growth, proliferation, and movement, are affected by the mutations suggesting that ESR1 mutations might cause similar phenotypes in breast and endometrial tumors. Motivated by the RNA-seq results, we measured proliferation and found that growth rates were not significantly different between mutant and wild-type clones; however, migration was significantly enhanced in the mutant clones. Consistent with the migration observations, we found that higher expression of several mutant-specific up-regulated genes and lower expression of several mutant-specific down-regulated genes in our data sets correlate with more aggressive tumors and poorer outcomes for endometrial cancer patients. Together our results indicate that ESR1 mutations have the potential to drive more dangerous forms of endometrial cancer, which is corroborated by a trend toward worse outcomes for patients with ESR1 mutant disease (Backes et al. 2016).
ESR1 mutations do not just confer ligand-independent estrogen signaling. In fact, most of the genes differentially expressed between the mutant lines and the wild-type lines are genes that do not normally respond to E2. This mutant-specific gene regulation appears to be directed by mutant ESR1 with the D538G mutation causing a large alteration in the genomic loci that ESR1 binds. Chromatin accessibility also increases at specific sites across the genome because of the D538G mutation, and a majority of these sites are bound by mutant ESR1. The increased chromatin accessibility at some mutant ESR1-bound sites could be the result of different underlying effects. One possibility is a small pioneering role for mutant ESR1, which could be attributable to changes in cofactor recruitment, as found in breast cancer (Jeselsohn et al. 2018), or because of its constant activity and binding. Another possibility is that ESR1 is taking advantage of the increased activity or expression of another transcription factor (e.g., NF-I factors) that has led to increased chromatin accessibility, which would represent an indirect effect of mutant ESR1 on the chromatin landscape.
To determine how much of mutant ESR1’s ability to regulate a new set of genes is related to its constant activity, we treated wild-type clones with continuous saturating doses of E2 for 25 d. Prolonged exposure to E2 changed the expression of thousands of genes and altered chromatin accessibility at thousands of loci; however, there was little overlap with the gene regulatory changes caused by the D538G mutation. These results suggest that the D538G ESR1 mutation is neomorphic/gain-of-function and does not simply cause hyperactivity. It is unclear how the mutation changes ESR1's gene regulatory role, but alterations to the placement of helix 12 of the ligand binding domain (Merenbakh-Lamin et al. 2013; Fanning et al. 2016) could cause changes in binding affinities to transcription factors or cofactors that bind to this region. Determining how mutant ESR1 causes novel gene regulation could provide valuable insights into treatment strategies aimed at blocking mutant ESR1's activity.
In this study, we focused on the D538G mutation because it is the only specific alteration in the LBD; L536 and Y537 have been found to be mutated to several different amino acids (Gaillard et al. 2019). In future studies, it will be interesting to determine if mutations to L536 and Y537 cause similar regulatory and phenotypic changes in endometrial cancer cells, because Y357S and D538G appear to cause mutation-specific alterations in breast cancer cells (Bahreini et al. 2017; Jeselsohn et al. 2018). In addition, our study focused on a particular endometrial cancer cell line, Ishikawa, and the effects of ESR1 mutations may be different in different models. We recently found that ESR1 genomic binding is consistent between endometrial tumors and distinct from breast tumors, with Ishikawa exhibiting a clear endometrial cancer ESR1 binding pattern (Rodriguez et al. 2019a), suggesting that our findings could be generally applicable. In summary, our study has led to the creation of isogenic models of mutant ESR1 in endometrial cancer cells, the confirmation of estrogen-independent mutant ESR1 activity, and the discovery of novel gene regulation through mutant ESR1 that cannot be explained by constant activity alone.
Methods
Plasmid construction for ESR1 LBD mutant generation
Mutant cell lines were created using the CETCH-seq method (Savic et al. 2015) in which a pFETCH plasmid is the homology donor (containing the mutation, 3× FLAG tag, P2A linker, and neomycin resistance gene) and Cas9 is targeted proximal to the stop codon by guide RNAs. To create the pFETCH homology donor plasmid with the D538G ESR1 LBD mutation, we used primers (for sequences, see Supplemental Table S3) to PCR amplify ESR1 homology arms, using a gBlock (IDT) (Supplemental Table S3) that encompassed 1000 bp surrounding the D538G mutation as a template for the amplification of homology arm 1 and genomic DNA from Ishikawa cells (Sigma-Aldrich) as the template for homology arm 2. For amplification, Phusion high-fidelity master mix (New England BioLabs) was used with 10 µM of each primer and 1 ng gBlock or 50 ng genomic DNA and amplified for 25 cycles. To generate the wild-type pFETCH donor plasmid, we repeated this technique with Ishikawa genomic DNA as a template for arm 1 amplification. Using BsaI and BbsI (New England BioLabs), we double digested the destination pFETCH vector (Addgene 63934, a gift from Eric Mendenhall and Richard M. Myers) and used Gibson assembly HiFi master mix (New England BioLabs) to clone homology arms 1 and 2 simultaneously to create D538G or wild-type pFETCH plasmids. Clones for each pFETCH vector underwent minipreps (Zymo Research) and were verified by Sanger sequencing (Genewiz). To create a Cas9 and guide RNA expressing plasmid that targeted near the stop codon of ESR1, a Cas9 and guide RNA expression vector (Addgene 62988, a gift from Feng Zhang) was digested with BbsI (New England BioLabs). Guide RNA oligos (Supplemental Table S3) were annealed and then ligated into the Cas9 guide RNA vector. Clones for each guide RNA underwent minipreps (Zymo Research) and were verified by Sanger sequencing (Genewiz). The pFETCH mutant and wild-type plasmids were SalI (New England BioLabs) digested before transfection to linearize the vector.
Cell culture and transfection for generation of ESR1 LBD mutant and wild-type lines
Ishikawa cells (Sigma-Aldrich) were seeded in six-well plates at a density of 300,000 cells per well in RPMI 1640 (Thermo Fisher Scientific) with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin-streptomycin (Thermo Fisher Scientific). At approximately 50% confluency, 250 ng of each of the two Cas9 guide RNA vectors and 250 ng of either the mutant or wild-type pFETCh vectors were transfected into cells with Lipofectamine 3000 (Thermo Fisher Scientific). Cells were also treated with 1 µM SCR7 (Xcessbio) to inhibit nonhomologous end joining (NHEJ) for 3 d post-transfection. Next, 72 h post-transfection, the media was changed, and G418 (Thermo Fisher Scientific) was added to a final concentration of 200 µg/mL. RPMI media and G418 were replaced every 2 d until resistant cells remained. To generate single-cell colonies, transfected cells were plated at limiting dilution and cultured until colonies were large enough to identify visually. Individual colonies were picked and transferred to a 24-well plate and grown until they reached confluency. At this time, genomic DNA was extracted from individual colonies using the DNeasy Blood and Tissue Kit (Qiagen). To genotype clones, we amplified a 700-bp region within the ligand binding domain of ESR1 using ESR1-LBD_F4 and ESR1-LBD_R1 primers (Supplemental Table S4). PCR products were purified with Ampure XP beads (Beckman Coulter) and Sanger sequenced (Genewiz) to confirm the D538G mutation or wild-type sequence (Supplemental Table S5). We also verified the presence of a tagged and untagged copy of ESR1 using ESR1-LBD_F4 and ESR1-LBD_STOP_R1 primers (Supplemental Table S4). The wild-type and ESR1 LBD mutant lines all harbored both a tagged and untagged copy of wild-type ESR1. Positive clones then underwent immunoblotting to verify protein expression.
Immunoblotting
Ishikawa ESR1 LBD mutant and wild-type cell lines were seeded in 100-mm dishes with RPMI 1640 with 10% fetal bovine serum and 1% penicillin-streptomycin until they reached confluency. At confluency, cells were washed with cold phosphate buffered saline (PBS), adherent cells were scraped, lysed with RIPA buffer (1× PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (Thermo Fisher Scientific), and sonicated with an Active Motif EpiShear probe-in sonicator with three cycles of 10 sec on, 10 sec of rest at 40% amplitude. Then 80 µg of protein was loaded onto a 4%–12% Bis-Tris gel (Thermo Fisher Scientific) and electrophoresed for 90 min at 130V in 1× MOPS buffer (Thermo Fisher Scientific). Proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane with the iBlot system (Thermo Fisher Scientific). Membranes were blocked in 5% milk/PBST for 1 h at room temperature. Blots were probed with primary antibodies to FLAG M2 (Sigma-Aldrich F1804) 1:1000, ESR1 HC-20 (Santa Cruz Biotechnology sc-543) 1:200, and actin beta C4 (Santa Cruz Biotechnology sc-47778) 1:1000 in 2.5% milk/PBST overnight at 4°C. Membranes were washed with PBST and incubated in secondary antibody (Goat anti-Mouse IgG, Thermo Fisher Scientific 32430; Goat anti-Rabbit IgG, Thermo Fisher Scientific 31460) in PBST at 1:5000 dilution. Protein signal was detected with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Cell culture
Ishikawa ESR1 LBD mutant and wild-type cells were cultured in full media: RPMI 1640 with 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were incubated at 37°C with 5% CO2 for the duration of all experiments. At least 5 d before inductions, cells were placed in hormone-deprived media: phenol-red free RPMI 1640 media (Thermo Fisher Scientific), because phenol-red is estrogenic, with 10% charcoal-dextran stripped fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin-streptomycin. Media was changed 1 d before inductions, and cells were treated with either DMSO (vehicle) or 10 nM E2 for 1 h for ChIP-seq and ATAC-seq experiments, or 8 h for RNA-seq and ATAC-seq experiments. For prolonged estrogen RNA-seq and ATAC-seq experiments, wild-type cell lines were cultured in phenol-red free RPMI with 10% charcoal-dextran stripped fetal bovine serum and 1% Penicillin-Streptomycin for up to 25 d. Cells were treated with 10 nM E2 every 2 d with media changes, and cell lysates were collected at the following time points: 8 h, day 10, day 15, day 20, and day 25.
ERE luciferase reporter assay
Approximately 15,000 cells per well for each cell line were seeded in a 96-well plate in hormone-deprived media for 5 d. Cells were then transfected, according to the manufacturer's protocol with inducible dual-luciferase and Renilla estrogen response element (ERE) constructs (Qiagen) using FuGENE HD transfection reagent (Promega). One day post-transfection, media was changed and cells were treated with either DMSO or 10 nM E2. Twenty-four hours post-treatment, luciferase activity was measured using the Dual-Glo luciferase assay system (Promega) per the manufacturer's instructions. All experiments were performed in individual wild-type and D538G mutant clones in triplicate with three biological replicates. Statistical analysis was performed using Student's t-test.
Quantitative PCR
Cell lysates were harvested following an 8 h E2 or DMSO control induction with buffer RLT plus (Qiagen) containing 1% beta-mercaptoethanol (Sigma-Aldrich). Total RNA was extracted and purified using a Quick-RNA Miniprep kit (Zymo Research). Twenty-five nanograms of RNA was used as starting material per sample, and qPCR was performed using the Power SYBR Green RNA-to-CT 1-Step Kit (Thermo Fisher Scientific) on a CFX Connect Real-Time light cycler (Bio-Rad). Primers for PGR, MMP17, EHF, EPHA3, and CTCF are listed in Supplemental Table S6. Expression measurements were calculated with the ΔΔCt method using CTCF as a control. Experiments were performed in all wild-type and D538G mutant clones in triplicate.
Proliferation and migration assays
Ishikawa ESR1 LBD mutant and wild-type cells were cultured in full media or hormone-deprived media for 3 d before plating for proliferation and migration experiments. For proliferation experiments, approximately 5000 cells per well for each cell line were seeded in 96-well plates in both media conditions. Cell proliferation was monitored via time-lapse image acquisition every 2 h, for up to 72 h, via the IncuCyte ZOOM imaging platform (Sartorius). Doubling times for individual cell lines in the two media conditions were calculated by performing linear regression between hours and the log base 2 confluency percentages that were normalized to the starting confluency. Doubling times were taken as 1 divided by the slope of the best fit line. Migration experiments were assessed via the wound healing assay. Approximately 30,000 wild-type and D538G mutant cells were seeded in 96-well ImageLock Microplates (Sartorius) and grown to confluency in full media and hormone-deprived media. The cell monolayer was scraped, and migration was monitored via time-lapse image acquisition every 2 h via the IncuCyte ZOOM imaging platform (Sartorius). Migration rates were calculated as relative wound density over time for wild-type and mutant cell lines. All experiments were performed in individual wild-type and D538G mutant clones in triplicate with three biological replicates. Statistical analysis was performed using Student's t-test.
ChIP-seq
Ishikawa ESR1 mutant and wild-type clonal cell lines were induced with DMSO or 10 nM E2 for 1 h, followed by fixation with 1% formaldehyde for 10 min at room temperature to cross-link cells. The cross-linking reaction was stopped with the addition of glycine to a final concentration of 125 mM. Cells were washed with cold PBS and harvested via cell scraping in Farnham lysis buffer supplemented with protease inhibitors. Chromatin immunoprecipitation was performed as previously described (Reddy et al. 2009) with an Anti-FLAG M2 (Sigma-Aldrich F1804) antibody. ChIP-seq libraries were sequenced on an Illumina HiSeq 2500, and sequencing reads were aligned to the hg19 build of the human genome using Bowtie (Langmead et al. 2009) with the following parameters: -m 1 -t --best -q -S -I 32 -e 80 -n 2. The hg19 build of the human genome was used for all genomic analyses. We do not believe that realigning reads to the current genome build (GRCh38) would substantially change results, because we are restricting our analyses to uniquely alignable regions of the genome. MACS2 (Zhang et al. 2008) was used to call peaks with a P-value cutoff of 1 × 10−10 and a mfold parameter between 15 and 100. Input control libraries from both wild-type and mutant cell lines were used as controls for each ChIP-seq experiment. All ChIP-seq experiments were performed in biological duplicates. Genomic annotation of binding sites was performed using CEAS (Ji et al. 2006). Overlaps between peaks were determined using a 1-bp minimum overlap, and percentages were calculated by dividing the number of overlapping peaks by the number of peaks in the smaller set (i.e., the percentage of maximal possible overlap). Differential binding sites were identified by using DESeq2 (Love et al. 2014) to compare counts per million at each ERBS that was identified in any ChIP-seq sample. ESR1 ChIP-seq experiments in wild-type clones treated with E2 were compared to all ESR1 ChIP-seq experiments performed in mutant clones. An adjusted P-value cutoff of 0.05 was used to identify mutant-enriched and wild-type–enriched ESR1-bound sites. Constant ERBS were regions bound by ESR1 in at least one replicate of the wild-type E2 FLAG ChIP-seq experiments and at least one replicate of the mutant FLAG ChIP-seq experiments but were not found to be differentially bound in the DESeq2 analysis. Motif finding was performed on 500-bp regions surrounding the summit of identified peaks. Motifs between 6 and 30 bp in length were identified by MEME Suite (Bailey et al. 2009), with a motif distribution of zero to one occurrence per sequence. To identify strong and weak EREs in mutant-specific and constant ESR1 binding sites, ERE scores were calculated by Patser (Hertz and Stormo 1999), within 100 bp of an individual peak's summit.
RNA-seq
Following 8-h treatments in hormone-deprived media with either 10 nM E2 or DMSO, cells were washed with PBS and harvested with buffer RLT plus (Qiagen) containing 1% beta-mercaptoethanol (Sigma-Aldrich). Cells were passed through a 21-gauge needle and syringe (Sigma-Aldrich) to lyse genomic DNA before RNA was extracted and purified using a Quick-RNA Miniprep kit (Zymo Research). Using the KAPA Stranded mRNA-Seq kit (KAPA Biosystems), poly(A) selected libraries were created with 500 ng of input RNA per sample. RNA-seq libraries were sequenced on an Illumina HiSeq 2500, and sequencing reads were aligned to the hg19 build of the genome using HISAT2 (Kim et al. 2015), with the University of California Santa Cruz (UCSC) Known Genes definitions used to build indexes. SAM files were converted to BAM files and sorted with SAMtools (Li et al. 2009). To quantify reads that mapped to UCSC Known Genes, we used featureCounts (Liao et al. 2014). Reads were normalized and analyzed for differential enrichment using the DESeq2 package in R (Love et al. 2014; R Core Team 2017). RNA-seq experiments were performed in each wild-type and D538G mutant clone, and clones with the same genotype were used as biological replicates in the analysis. To parse differentially regulated gene lists, we first identified statistically significant genes (adjusted P-value <0.05) that were up-regulated and down-regulated in response to an E2 induction in wild-type cells. To find genes that are regulated by E2 in wild-type clones and change expression in the mutant clones without E2, we used DESeq2 to compare wild-type DMSO-treated samples to mutant DMSO-treated samples and compared the genes to E2 regulated genes in wild-type clones. To identify novel genes not normally regulated by E2, we used DESeq2 to compare all D538G samples to all wild-type samples treated with E2 and then subtracted genes that were regulated by E2 in wild-type cells. The adjusted P-value cutoff for all significant genes in each comparison was <0.05. Differentially expressed novel up-regulated and down-regulated genes were analyzed through the use of Ingenuity Pathway Analysis (Qiagen; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis). The five statistically significant molecular and cellular functions identified in this analysis are included in Supplemental Figure S1B,C.
For prolonged RNA-seq experiments, ESR1 wild-type cell lines were treated with 10 nM E2 every 2 d along with media changes, and cell lysates were collected at the following time points: day 10, day 15, day 20, and day 25. Total RNA was extracted, and poly(A) selected libraries were constructed, sequenced, and analyzed as described above. The two wild-type clones were used as biological replicates in this analysis. To identify genes differentially regulated in response to prolonged E2, we compared a group consisting of the DMSO-treated and 8-h E2-treated samples to a group consisting of the 10-, 15-, 20-, and 25-d samples. For DESeq2 analysis, the two groups were treated as categorical variables. The adjusted P-value cutoff for all significant genes was <0.05.
TCGA data analysis
RNA-seq and clinical data was obtained from the TCGA data portal in December 2015. The gene expression measurements used were level 3 RNaseqV2 normalized RSEM data. Only samples with endometrioid histology and ESR1 expression above the median among the endometrioid tumors were analyzed for survival analysis. Cox regression was used to evaluate the association between gene expression and progression-free survival in R using coxph from the survival package. We used a median cutoff to define high and low expressing tumors for each gene when running the Cox regression.
ATAC-seq
After 1- and 8-h treatments with either 10 nM E2 or DMSO, cells were trypsinized and isolated by centrifugation. Next, 250,000 cells were isolated from ESR1 LBD wild-type or mutant cell lines, and ATAC-seq was performed as previously described (Buenrostro et al. 2013). ATAC-seq libraries were sequenced on an Illumina HiSeq 2500, and sequencing reads were aligned to hg19 using Bowtie (Langmead et al. 2009) with the following parameters: -m 1 -t --best -q -S -I 32 -e 80 -n 2. SAM files were converted to BAM files and sorted with SAMtools (Li et al. 2009). MACS2 (Zhang et al. 2008) was used to call peaks without an input control but with a P-value cutoff of 1×10−10. We used featureCounts (Liao et al. 2014) to quantify reads that aligned to all ATAC-seq peaks called in any sample. Reads were normalized and analyzed for differential enrichment using the DESeq2 package for R (Love et al. 2014). ATAC-seq experiments were performed in each wild-type and D538G mutant clone, and clones with the same genotype were used as biological replicates. Mutant-enriched and mutant-depleted ATAC-seq sites were identified by comparing ATAC-seq signal between all wild-type and all mutant samples. Motif discovery at ESR1-associated and non-ESR1-associated regions was performed on 500-bp regions surrounding the summit of identified peaks. Motifs between 6 and 30 bp in length were identified by MEME Suite (Bailey et al. 2009), with a motif distribution of zero to one occurrence per sequence.
For prolonged ATAC-seq experiments, ESR1 wild-type clones were treated with 10 nM E2 every 2 d during media changes, and cells were collected at the following time points: day 10, day 15, day 20, and day 25. ATAC-seq experiments and analysis were performed as described above. Prolonged E2-enriched and prolonged E2-depleted ATAC-seq regions were identified by comparing ATAC-seq signal between all wild-type samples treated with DMSO or E2 for 1 h to all wild-type samples exposed to prolonged E2 (10, 15, 20, and 25 d). For DESeq2 analysis, the two groups were treated as categorical variables. The adjusted P-value cutoff for all significant genes was <0.05. Genomic annotation of these regions was performed using CEAS (Ji et al. 2006).
Statistical and graphical packages
The statistical analyses were performed in R version 3.3.2 (R Core Team 2017), except for the P-values for novel gene enrichments calculated by IPA and P-values for enriched motifs calculated by MEME. Heatmaps were generated in R using the pheatmap package, and statistical tests used for analysis and corresponding P-values can be found throughout the text. Heatmaps for ChIP-seq and ATAC-seq data were generated by displaying the Z-score across a region based on the reads per million that aligned to each region in each sample.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the following accession numbers: GSE132428 (RNA-seq), GSE132426 (ChIP-seq), and GSE132424 (ATAC-seq).
Acknowledgments
This work was supported by a Department of Defense Breast Cancer Research Program Breakthrough Award to J.G. (W81XWH-16-1-0421) and the Huntsman Cancer Institute. Research reported in this publication used the High-Throughput Genomics Shared Resource at the University of Utah and was supported by National Institutes of Health/National Cancer Institute award P30 CA042014. We thank Ed Grow for providing reagents, and Margit Janat-Amsbury, Jennifer Richer, K.-T. Varley, as well as Gertz and Varley laboratory members for their helpful comments on the study and the manuscript.
Author contributions: The project was conceptualized by Z.B. and J.G., and the methodology was designed by Z.B. and J.G. Investigation was performed by Z.B., K.C.B., S.A., and J.G., and formal analysis was done by Z.B., J.M.V., and J.G. The original draft was written by Z.B. and J.G.; and Z.B., J.M.V., K.C.B., S.A., and J.G. reviewed and edited the manuscript. J.G. acquired the funding.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.244780.118.
- Received October 2, 2018.
- Accepted July 23, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








