
Pervasive and dynamic transcription initiation in Saccharomyces cerevisiae
Abstract
Transcription initiation is finely regulated to ensure proper expression and function of genes. The regulated transcription initiation in response to various environmental stimuli in a classic model organism Saccharomyces cerevisiae has not been systematically investigated. In this study, we generated quantitative maps of transcription start sites (TSSs) at a single-nucleotide resolution for S. cerevisiae grown in nine different conditions using no-amplification nontagging Cap analysis of gene expression (nAnT-iCAGE) sequencing. We mapped ∼1 million well-supported TSSs, suggesting highly pervasive transcription initiation in the compact genome of the budding yeast. The comprehensive TSS maps allowed us to identify core promoters for ∼96% verified protein-coding genes. We corrected misannotation of translation start codon for 122 genes and suggested an alternative start codon for 57 genes. We found that 56% of yeast genes are controlled by multiple core promoters, and alternative core promoter usage by a gene is widespread in response to changing environments. Most core promoter shifts are coupled with altered gene expression, indicating that alternative core promoter usage might play an important role in controlling gene transcriptional activities. Based on their activities in responding to environmental cues, we divided core promoters into constitutive class (55%) and inducible class (45%). The two classes of core promoters display distinctive patterns in transcriptional abundance, chromatin structure, promoter shape, and sequence context. In summary, our study improved the annotation of the yeast genome and demonstrated a much more pervasive and dynamic nature of transcription initiation in yeast than previously recognized.
The RNA polymerase II (Pol II) core promoter is the region where Pol II is recruited to initiate transcription. It includes the transcription start sites (TSSs) and immediately flanking sequences that contain various DNA motifs to accurately direct transcription initiation by the preinitiation complex (PIC) (Butler and Kadonaga 2002). The core promoter is the final target of actions of almost all the factors involved in transcriptional regulation because regulatory signals of transcription are ultimately integrated into the initiation process at core promoters (Juven-Gershon and Kadonaga 2010). Accurate transcription initiation is vital to ensure proper expression and function of genes (Smale and Kadonaga 2003). Misregulation of transcription initiation has been found to be associated with a broad range of human diseases, such as breast cancer, diabetes, kidney failure, and Alzheimer's disease (Arrick et al. 1991; Romeo et al. 1993; Capoulade et al. 2001; Sobczak and Krzyzosiak 2002; Mihailovich et al. 2007). In this regard, accurate identification of TSSs and characterization of their regulated activities are essential for obtaining fundamental insights into regulatory mechanisms that determine the location and activities of transcription initiation. The global maps of TSSs and core promoters have been generated in several important metazoans, such as human (The ENCODE Project Consortium 2012), mouse (The FANTOM Consortium 2005), fruit fly (Hoskins et al. 2011), and zebrafish (Haberle et al. 2015). These maps revealed that most animal genes contain multiple core promoters, and the selections and activities of core promoters are precisely regulated to ensure that a correct transcript is produced at an appropriate level in a tissue or developmental stage (Carninci et al. 2006; The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014).
The budding yeast Saccharomyces cerevisiae has served as a classic model organism for many landmark discoveries in gene regulation and other cellular processes over the past several decades (Duina et al. 2014). Various techniques have been applied to identify genome-wide TSSs in S. cerevisiae, such as microarray (Hurowitz and Brown 2003; David et al. 2006; Xu et al. 2009), serial analysis of gene expression (SAGE) (Zhang and Dietrich 2005), sequencing of full-length cDNA clones (Miura et al. 2006), RNA sequencing (Nagalakshmi et al. 2008; Waern and Snyder 2013), Transcript isoform sequencing (TIF-seq) (Pelechano et al. 2013; Park et al. 2014), transcript-leaders sequencing (TL-seq) (Arribere and Gilbert 2013), and TSS-seq (Malabat et al. 2015). Tuning gene expression is an essential way to maximize cell survival through rapid responses to environmental stresses, particularly for unicellular organisms (de Nadal et al. 2011). Therefore, identification and characterization of the TSS activities in response to changing environments is important for gaining fundamental insights into regulatory mechanisms of transcription initiation in yeast. Previous studies based on TIF-seq, TL-seq, and TSS-seq were performed in only one or two growth conditions (Arribere and Gilbert 2013; Pelechano et al. 2014; Malabat et al. 2015). The TSSs and core promoters identified in these studies may only represent a small portion of the transcription initiation landscape in yeast, because many transcripts are generated only in particular growth conditions. It is also challenging to accurately characterize the dynamic activities of core promoters with one or two examined conditions. In another study, RNA sequencing was carried out in S. cerevisiae grown in 18 different conditions and extensive different 5′ ends have been observed, suggesting the dynamic of transcription initiation in yeast under different growth conditions (Waern and Snyder 2013). However, RNA-seq is known to have a shortcoming of inaccurate determination of TSSs, as assembly of RNA-seq reads usually extends transcript contigs to the very 5′ end, which lack the information of other TSSs within the longest transcript (Batut et al. 2013; Steijger et al. 2013; Boley et al. 2014; Wade and Grainger 2014). Therefore, it is necessary to generate high-resolution and quantitative TSS maps for yeast cells grown under various conditions to better understand the regulated activities of transcription initiation.
A revised Cap analysis gene expression (CAGE) technique, called no-amplification nontagging CAGE libraries for Illumina sequencers (nAnT-iCAGE), is ideal for generating TSS maps at a single-nucleotide resolution and simultaneously quantifying their activities (Murata et al. 2014). Similar to TIF-seq (Pelechano et al. 2013) and TSS-seq (Malabat et al. 2015), nAnT-iCAGE captures the 7-methylguanosine cap structure at the 5′ end of transcripts and sequences the transcripts using high-throughput sequencers. By mapping sequenced reads to a reference genome, the exact TSS locations can be identified. The number of reads mapped to a TSS also quantifies the number of transcripts initiated at the TSS. Moreover, nAnT-iCAGE does not involve PCR amplification or restriction enzyme digestion, which reduces bias on the transcription level owing to sequence-dependent efficiency of PCR and loss of RNA samples caused by restriction enzyme digestion (Murata et al. 2014). In this study, we generated quantitative TSS maps for S. cerevisiae grown under nine different conditions with nAnT-iCAGE and characterized its regulatory dynamics of transcription initiation in an unprecedented depth and breadth.
Results
Pervasive transcription initiation in S. cerevisiae
The S. cerevisiae strain BY4741, a haploid derivative of the laboratory strain S288c, was used as the study system to generate high-resolution TSS maps. The 5′ boundaries of transcripts were captured following the nAnT-iCAGE protocol from S. cerevisiae cells grown in nine conditions (Table 1), which are informative on the natural environments and common stresses of wild yeast populations. For each growth condition, two biological replicates of nAnT-iCAGE libraries were constructed (18 libraries in total). All nAnT-iCAGE libraries were sequenced using Illumina NextSeq 500 (single-end, 75-bp reads), which yielded 636 million sequencing tags in total (Supplemental Table S1), providing an unprecedented depth of coverage for 5′ boundaries of transcripts in yeast.
Growth conditions examined in this study
With a mapping rate of 91.9%, 584,689,028 tags were aligned to the reference genome of S. cerevisiae (assembly R64-2-1). We only used tags that are uniquely mapped to the reference genome (348,493,668 tags) for further analysis. The Pearson correlation coefficient r of the tag counts of the CAGE tags identified TSS (CTSSs) between two biological replicates of each growth condition range from 0.97 to 1 (Supplemental Fig. S1), supporting the high reproducibility of the nAnT-iCAGE technique. Systematic G nucleotide addition bias at the 5′ end of CAGE tags was corrected based on the probability of G addition (Carninci et al. 2006). The numbers of CTSSs identified in each growth condition range from 1,106,287 to 1,632,079 (Supplemental Table S2). We mapped 4,254,561 unique CTSSs by combining data from all samples. However, 52.8% of CTSSs are only supported by 1–2 uniquely mapped tags (Supplemental Fig. S2). These CTSSs could be caused by technical artifacts or the stochastic transcription, which is the main source of significant cell-to-cell variations at mRNA levels (Raj and van Oudenaarden 2008). To minimize the false CTSSs, we only considered those with tags per million (TPM) ≥0.1 for further analysis (on average, supported by at least three uniquely mapped tags). The numbers of eligible CTSSs range from 315,546 to 511,937 (median = 395,182) across the nine samples. Combination of CTSSs obtained from all growth conditions yielded 925,804 unique CTSSs, which doubles the number of CTSSs identified by any single growth condition (Fig. 1A; Supplemental Table S2), supporting that it is necessary to examine more growth conditions to obtain a more complete TSS landscape in yeast. Although we used a conservative threshold, the number of TSSs identified in this study is about four times of previous studies in S. cerevisiae. Specifically, based on 1.88 million tags generated by TIF-seq from two growth conditions (Pelechano et al. 2013), and 8.6 million tags generated by TL-seq from one growth condition (Arribere and Gilbert 2013), the numbers of TSS supported by at least two tags are 227,021 and 204,197, respectively (Supplemental Fig. S2). TSS-seq data mapped 225,563 TSSs supported by TPM ≥ 0.1 (Malabat et al. 2015). In the fission yeast Schizosaccharomyces pombe, which has a similar genome size (12.61 Mb) as S. cerevisiae, only 93,736 CTSSs were supported by a single CAGE tag (Li et al. 2015). Therefore, by increasing the depth and breadth of TSS profiling, our results support that the transcription initiation in the unicellular yeast is much more pervasive than previously recognized.
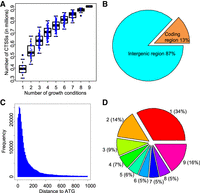
Pervasive and dynamic transcription initiation in S. cerevisiae. (A) Correlations between numbers of examined growth conditions and identified CTSSs. The x-axis indicates the number of examined conditions. Each dot in the box plots represents the number of identified CTSSs based on a combination of TSS data from N numbers (ranging from 1 to 9) of growth conditions. (B) Distribution of mapped CAGE tags in different genomic regions. (C) Distribution of distance between CTSSs and annotated translation start codon (ATG). (D) Proportions of CTSSs identified in different numbers of growth conditions.
As expected, most CAGE tags (87%) were mapped to the intergenic regions (Fig. 1B), supporting that most transcription is initiated from noncoding regions. The CTSSs are highly enriched within 200 bp upstream of annotated translation start codon. The distribution of CAGE tags forms a sharp peak at ∼30–40 nt upstream of the start codon (Fig. 1C). Therefore, the most common size of the 5′ untranslated region (5′ UTR) of mRNA transcripts in yeast is ∼30 nt, which is probably the optimal size for binding of 40S ribosomes. It is worth noting that only a small portion of CTSSs (16%) have detectable activity in all conditions examined. In contrast, transcription activities from 34% of them can only be detected in one growth condition (Fig. 1D), suggesting a highly dynamic activity of TSS in response to environmental cues.
Identification of core promoters and improvement of genome annotation
The core promoter was typically defined as a stretch of contiguous DNA sequence encompassing the TSSs (Butler and Kadonaga 2002). The availability of multiple quantitative TSS maps allowed us to generate a more complete and accurate map of core promoters in yeast. We used a hierarchical approach to infer core promoters by integrating the TSS maps obtained from all growth conditions (Methods). A total number of 43,325 consensus clusters were inferred based on the nine TSS maps, representing the largest number of putative core promoters identified in yeast so far. We then assigned the consensus clusters to Pol II transcribed genes as their core promoters based on the distance between their dominant TSS (the CTSS with highest TPM) and the annotated boundaries of downstream genes (Supplemental Fig. S3; Supplemental Methods).
We noticed that many consensus clusters locate downstream from annotated translation start codons or intragenic regions. These consensus clusters were generally of lower abundance, which is consistent with previous studies based on other techniques (Miura et al. 2006; Arribere and Gilbert 2013; Malabat et al. 2015). Given the pervasive nature of transcription in eukaryotes, they could be cryptic promoters within gene bodies that provoke spurious intragenic transcription (Kaplan et al. 2003). Another possibility is the misannotation of translation start codons (Cliften et al. 2003; Kellis et al. 2003) or the presence of alternative translation start codons (Bazykin and Kochetov 2011). We manually examined the intragenic consensus clusters to identify misannotations or alternative translation start codons (Fig. 2A–C). Because translation is generally initiated at the first AUG codon encountered by ribosomes during scanning of mRNA in the 5′-to-3′ direction (Kozak 2005), we searched for the first in-frame ATG codon downstream from the intragenic consensus clusters, which is likely the correct or alternative translation start codon of a gene.
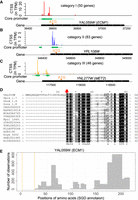
TSS maps improve yeast genome annotation. (A–C) Examples of CAGE signal distribution in Category I, II, and III genes. In each example, the top track illustrates the distributions of CTSS signals near the annotated ORF. The middle track (green box) represents the core promoter region. The vertical line represents the dominant TSS in each core promoter. The bottom track displays the locations of annotated ORF. The originally annotated ATG is at the far side of the black box (ORF). Revised start codon (A,B) or alternative start codon (C) are indicated by “ATG” and an orange triangle. (D) Multiple sequence alignment for orthologous protein sequences of YAL059W (ECM1). Only the first 60 alignment sites are shown. The first 24 amino acids in the N terminus of YAL059W are absent in its orthologous sequences. The red arrow indicates the revised start codon. (E) A histogram shows the number of observations for detected peptides in ECM1. No peptides have been detected by previous mass spectrometry studies in the section between two orange lines, which corresponds to the 24 amino acids of the misannotated region in YAL059W.
Based on the presence, location, and transcriptional abundance of intergenic and intragenic consensus clusters, we first identified three categories of genes (I, II, and III) representing different degrees of likelihood of being misannotated or having alternative start codons (Supplemental Methods). The Category I (50 genes) is the group with the highest likelihood of misannotation, because they do not have any upstream core promoters, but have at least one intragenic consensus cluster near their 5′ end of ORFs (Fig. 2A; Supplemental Table S3). The Category II group (80 genes) have both intergenic and intragenic consensus clusters, but the intragenic ones have a stronger transcript abundance (Fig. 2B; Supplemental Table S4). The Category II genes are likely misannotated, but we cannot exclude the possibility that a downstream in-frame ATG codon is used as their alternative start codon. The Category III (46 genes) also have both intergenic and intragenic consensus clusters, but they have similar transcript abundance. Thus, multiple translation start codons may be used, but misannotation is still a possibility (Fig. 2C; Supplemental Table S5).
We then integrated other types of evidence to facilitate revisions of open reading frame (ORF) annotations for genes in the three categories. The TSS profiling data from other studies, including TIF-seq (Pelechano et al. 2013), TL-seq (Arribere and Gilbert 2013), and TSS-seq (Malabat et al. 2015), supported that majority of intragenic TSS clusters identified by this study were also observed by at least one of the three techniques (Supplemental Methods; Supplemental Data S1). For each candidate gene, we examined whether the potentially misannotated regions are absent or divergent in their orthologous sequences obtained from the Yeast Gene Order Browser (YGOB) (Supplemental Methods; Supplemental Data S2; Byrne and Wolfe 2005). We also determined whether peptides have been detected in the potentially misannotated regions based on a large collection of proteomic data compiled by the Global Proteome Machine Database (GPMDB) (Supplemental Methods; Supplemental Data S3; Craig et al. 2004). For instance, the misannotated region of YAL059W is absent in the sequences of its orthologous proteins (Fig. 2D), and its peptides have not been detected in S. cerevisiae (Fig. 2E). Integrative analyses of these data resulted in correction of translation start codon for 122 genes and suggestion of alternative start codon for 57 genes (Supplemental Table S3–S5). Based on our revisions, we generated an updated genome annotation file in General Feature Format (GFF) format for S. cerevisiae (Supplemental Data S4).
Based on the revised ORF annotations, we have assigned 11,462 consensus clusters to 5954 ORFs as their core promoters, including 5554 verified ORFs and 400 dubious ORFs. Therefore, our study inferred core promoters for 95.8% of 5797 verified protein-coding genes (assembly R64-21-1). These ORF-associated consensus clusters contain 88.5% of uniquely mapped tags. We also assigned 255 consensus clusters to 92 noncoding genes, such as snRNAs and snoRNAs, accounting for 4.9% of uniquely mapped tags (Supplemental Data S5). Furthermore, a total number of 555, 1944, and 370 consensus clusters were assigned to the predicted CUTs, XUTs, and SUTs, respectively (Xu et al. 2009; van Dijk et al. 2011). The remaining 28,741 consensus clusters are not associated with any Pol II transcribed genes based on our criteria (Supplemental Data S5). Most of these unassigned clusters have low transcriptional activity and only include 5.7% of uniquely mapped tags. The presence of abundant low-activity consensus clusters might reflect the prevalent stochastic or cryptic transcription initiation in the yeast genome. However, we cannot exclude the possibility that some of them could be the core promoters of unknown Pol II transcribed genes. Our consensus cluster data would be valuable for future studies to identify novel genes or transcripts in yeast.
Dynamic activity of core promoters in response to environmental cues
Our comparative analysis of TSS maps unraveled a highly dynamic landscape of transcription initiation in the yeast genome in response to changing environments. To better characterize the functional significance of dynamic transcription initiation, we divided the yeast protein-coding genes into two groups based on the number of assigned core promoters. If a gene is controlled by a single core promoter, it was classified as a single-core-promoter gene, which includes 44% of yeast genes (Supplemental Fig. S4; Supplemental Data S6). The remaining genes (56%) are controlled by two or more core promoters, which were classified as multi-core-promoter genes. The proportion of multi-core-promoter genes in yeast is similar to human (58%) (Carninci et al. 2006). Unlike the human genome, the S. cerevisiae genome is highly compact, and only 30% of the yeast genome are intergenic regions. The average length of the intergenic sequence in yeast is much shorter than that of the human genome (2.2 kb vs. 71 kb). Therefore, despite the huge discrepancy in the size of intergenic sequences, the proportions of multi-core-promoter genes are similar between human and yeast.
We noticed that in many multi-core-promoter genes, the distributions of CAGE signals between core promoters have significantly changed in response to environmental cues, suggesting alternative usage of core promoters, or core promoter shift (Methods; Supplemental Table S5). For instance, the CIK1 gene (YMR198W), which encodes kinesin-associated proteins involved in controlling both the mitotic spindle and nuclear fusion during mating (Page and Snyder 1992; Shanks et al. 2001), has two core promoters located ∼300 nt from each other. Transcription of CIK1 in yeast cells grown in rich medium (YPD) is exclusively initiated from the distal core promoter (Fig. 3A). Upon exposure to the mating pheromone α factor (cell arrest), almost all transcription initiation of CIK1 switched to the proximal core promoter (Fig. 3A). Our experimental validation supported the presence of two core promoters in CIK1 and their activity shift in response to α factor treatment (Fig. 3B). The similar observations in two other genes, YCR089W (FIG2) and YER065C (ICL1), were also supported by our experimental validations (Supplemental Fig. S5).
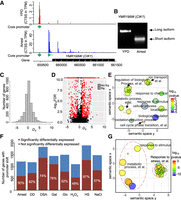
Prevalent core promoter shift responding to changing environments. (A) An example of core promoter shift (CIK1) between two growth conditions, YPD and arrest. (B) Experimental validation displays the presence and shift of two CIK1 transcript isoforms in response to changing environments. (C) Distribution of “degree of shift” (Ds) values in “H2O2” growth condition, using “YPD” as a control. (D) Volcano plot displays the correlations between Ds and −log10 FDR values (χ2 test). Each dot represents one gene. Dots in red represent genes with significant promoter shift (FDR < 0.05 and Ds < −1 or Ds > 1). (E) Scatterplot of enriched GO terms with significant core promoter shift (FDR < 0.05 and Ds < −1 or Ds > 1) under H2O2 condition. (F) The proportions of genes with core promoter shift that also experienced differential gene expression in response to environmental cues (FDR < 0.05, DESeq2). The percentages are indicated in each bar. (G) Scatterplot of enriched GO terms with significant core promoter shift and altered gene expression.
To determine the prevalence of core promoter shift, we calculated “degree of shift” (Ds) values based on the changes of CAGE tag distribution between core promoters for each multi-core-promoter gene (Methods). We found that 2833 of 3349 (85.6%) multi-core-promoter genes have experienced a significant shift of core promoter activity in at least one condition (FDR < 0.05, χ2 test), demonstrating widespread core promoter shifts in response to changing environments in yeast (Supplemental Data S7). The distribution of Ds values approximately followed the normal distributions (Fig. 3C), indicating that many factors may impact the selection of core promoters in different growth conditions. Gene Ontology (GO) enrichment analyses of genes with significant core promoter shift (Ds < −1 or Ds > 1, FDR < 0.05) (Fig. 3D) demonstrated that these genes are overrepresented in the groups with functions related to the adaptation to environmental stimuli, supporting a functional significance of core promoter shift (Fig. 3E; Supplemental Fig. S6).
We found that core promoter shifts responding to changing environments tend to be coupled with gene differential expression. In the case of CIK1, its transcription level is significantly up-regulated from 12.8 TPM in YPD to 200.3 TPM after α factor treatment. The up-regulation of CIK1 in response to α factor treatment is consistent with a previous study based on northern blot (Kurihara et al. 1996). The switch of core promoters by CIK1 is also supported by a low-throughput study using 5′ RACE and the shift produced a shorter Cik1p isoform that lacks 34 AA at the N terminus (Benanti et al. 2009). Because the N terminus of Cik1p is important for its nuclear localization and it contains a sequence that is necessary for ubiquitination, the shorter Cik1p isoform is more stable. Therefore, this case suggests that core promoter shift may have a significant impact on gene expression and protein function.
Depending on growth conditions, 48.9%–76.1% of genes with core promoter shift are coupled with significant gene differential expression (Fig. 3F; Supplemental Data S8). Therefore, similar to the regulated alternative usage of core promoters in different tissues in human and mouse, condition-specific transcripts are commonly generated by alternative usage of core promoters in unicellular eukaryotic organisms. GO analysis of genes with both promoter shift and differential expression showed that these genes are enriched in the groups related to the specific stress conditions (Fig. 3G; Supplemental Fig. S7). Thus, our observations suggested that core promoter shift might function as a mechanism for fine-tuning of gene expression and the adaptation to changing environments in yeast.
Two classes of core promoters in S. cerevisiae
The regulated transcription initiation was mostly characterized at the gene level in previous studies (Tirosh and Barkai 2008; Wu and Li 2010; Rosin et al. 2012). Considering that most genes are controlled by multiple core promoters and the alternative usage of core promoters is prevalent, it is more informative to characterize the dynamic activity of transcription initiation at the core promoter level. Among the 11,462 core promoters assigned to protein-coding genes, only 55% of them (6251) have detectable transcriptional activities under all examined conditions (Fig. 4A). The transcriptional activities of 17% of core promoters can only be detected in one growth condition, suggesting a strong condition specificity (Fig. 4A). Based on their transcription activities across nine growth conditions, we classified yeast core promoters into two classes: constitutive core promoter and inducible core promoter (Supplemental Data S6). If transcription initiation constitutively occurs from a core promoter in all examined growth conditions, we defined it as a constitutive core promoter. Only 5211 (45%) core promoters belong to the constitutive class. If the transcriptional activity of a core promoter can only be detected in one or some of the examined growth conditions, it was classified as an inducible core promoter, which accounts for 55% of all core promoters (Fig. 4A).
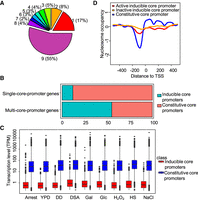
Distinct properties of constitutive and inducible core promoters in S. cerevisiae. (A) Pie chart shows the fractions of core promoters that can be detected in different numbers of growth conditions. Numbers in the pie chart represent the numbers of growth conditions. (B) Core promoter compositions in single- and multi-core-promoter genes. (C) Transcript abundance of constitutive core promoters and inducible core promoters in all examined growth conditions. (D) Patterns of nucleosome occupancy around dominant TSSs of different types of core promoters. The nucleotide occupancy data were obtained in rich medium (YPD).
The distributions of the two classes of core promoters are significantly different between the single-core-promoter genes and multi-core-promoter genes (Fig. 4B; Supplemental Table S2). Specifically, 88% of core promoters in single-core promoter genes are constitutive core promoters, whereas only 45% of core promoters in multi-core-promoter genes are constitutive core promoters. This observation suggests that most single-core-promoter genes tend to be constitutively expressed in yeast regardless of growth environments. In contrast, in the multi-core-promoter genes, most of their core promoters are only active under specific conditions. Based on our GO enrichment analysis (Supplemental Fig. S8), multi-core-promoter genes are significantly enriched in gene regulation processes, such as regulation of biological process, regulation of cellular process, biological regulation, and so forth.
Another distinct pattern between the two types of core promoters is their transcriptional abundance. At a genome-scale, the transcriptional activity from constitutive core promoters is significantly stronger than that of inducible core promoters in all examined conditions (Fig. 4C). We speculated that the different transcriptional abundance between the two types of core promoters is caused by different nucleosome positioning patterns, which were shown to have major impacts on transcriptional activity (Jiang and Pugh 2009; Nocetti and Whitehouse 2016). The eukaryotic DNA is coiled around a core of histones that form nucleosomes. If nucleosomes are present in the core promoter region, it becomes an obstacle for transcription initiation. Thus, the activation of transcription from such core promoters requires alteration of chromatin structure by ATP-dependent nucleosome sliding (Shen et al. 2000; True et al. 2016) or histone modification (Shilatifard 2006). To test this hypothesis, we compared the chromatin structure between the two classes of core promoters (± 500 nt of dominant TSS) using the nucleosome occupancy data obtained from Field et al. (2009). We observed a nucleosome-free region (NRF) immediately upstream of TSS in constitutive core promoters (Fig. 4D; Supplemental Fig. S9). In contrast, the inducible core promoters generally are occupied with nucleosomes in the same region. Most inducible core promoters are inactive in yeast cells grown in YPD. Consistently, we observed a more depleted nucleosome occupancy upstream of TSSs in the active inducible core promoters than that of the inactive ones (Fig. 4D), supporting that nucleosome occupancy pattern plays a determining role in controlling the transcriptional activity of a core promoter. Therefore, because of the difference in chromatin structure, different mechanisms are likely involved in transcription activation of the two types of core promoters. It was proposed that nucleosome positioning is largely determined by the intrinsic property of nearby DNA sequences (Kaplan et al. 2009; Tirosh et al. 2010). It is possible that the different genomic context might underlie the distinct chromatin structures between the two types of core promoters.
Distinct promoter shape between inducible and constitutive core promoters
Transcription can be initiated at precise positions or a disperse region, which form a continuum of shape of core promoters from sharp to broad shape (Carninci et al. 2006; Hoskins et al. 2011). The promoter shape is generally conserved between different species (Carninci et al. 2006; Main et al. 2013), and different signatures of evolution have been observed between broad and sharp core promoters (Schor et al. 2017), supporting important but distinct functional roles between promoters with different shapes. Similar to metazoan species, the spatial distribution of CAGE signals varies substantially among core promoters in S. cerevisiae, spanning a range of shapes from peaked to broad (Fig. 5A). To characterize the shape of yeast core promoters and to determine the extent to which inducible core promoters differ from constitutive core promoters in promoter shape, we developed a new metric to describe promoter shape, called Promoter Shape Score (PSS) (Methods). PSS integrates the observed probability of tags at each TSS within a core promoter and its quantile width. The main improvement of PSS over previous promoter shape estimation method Shape Index (SI) (Hoskins et al. 2011) is that SI does not take into consideration the distances between TSSs, which determine the promoter width. Without integrating promoter width factor, SI does not distinguish the difference between two promoters if tags are discontinuously distributed.
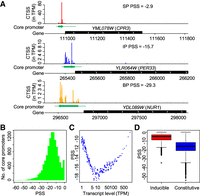
Classifications of core promoter shape. (A) Examples of sharp, intermediate, and broad core promoters in S. cerevisiae. Core promoters with PSS greater than −10 were classified as sharp core promoters (SP), smaller than −20 as broad core promoters (BP), and the others as intermediate core promoters (IP). (B) Histogram shows the distribution of PSS values in S. cerevisiae. (C) Relationships between the transcript abundance and PSS values of core promoters. The dot plot was generated using a sliding window analysis after sorting all core promoters by transcript abundance (TPM). Each window has 200 core promoters with a moving step of 40 core promoters. Each dot presents the median values of PSS and TPM of each window. (D) A box plot of PSS values of inducible core promoters and constitutive core promoters.
Based on our algorithm of PSS, the sharpest core promoter has a PSS value of 0, which means that all transcription initiation of a core promoter occurs from a single TSS (also called singleton). The PSS value decreases when the number of TSSs increases and/or the distribution of transcription initiation signals becomes more even. The PSS values of all core promoters of protein-coding genes largely follow a Gaussian distribution (−14.8 ± 8.07). We noticed that there are more core promoters with a PSS close to 0 than expected numbers based on Gaussian distribution (Fig. 5B). This is a result of the presence of many singleton core promoters. As shown in Figure 5B, PSS values form a peak in the range of −20 to −10. Therefore, based on PSS values, we classified core promoters into three groups: sharp core promoter (SP) with PSS > −10; broad core promoters (BP) with PSS ≤ −20; and the rest are considered as intermediate-shaped core promoters (IP) (Fig. 5A).
To determine the relationships between core promoter shape and gene expression patterns, we conducted a sliding window analysis between PSS and their transcript abundance. By plotting the median PSS and TPM values of each window of 200 core promoters, we observed a “V” shape between the two metrics (Fig. 5C). In general, for core promoters with transcript abundance < 10 TPM, the core promoters become broader with an increase of transcript abundance. In core promoters with the lowest activity, transcription is usually initiated from a single TSS, which form an ultrasharp promoter (PSS = 0). The increase of transcription activity from these low-activity core promoters appears to be mainly achieved by expanding TSSs, resulting in a broader promoter shape. However, for core promoters with transcript abundance > 10 TPM, the core promoters become sharper with an increase of transcript abundance. This observation suggests that the increase of transcript abundance is mainly achieved by increased transcription from one or a few TSSs within these core promoters, rather than expanding TSSs, which forms a positive correlation between transcription activity and PSS.
The PSS values of constitutive core promoters are significantly lower than that of inducible core promoters (P < 2.2 × 10−16, Student's t-test) (Fig. 5D). Of 5211 inducible core promoters, 4267 of them were classified as SP, and only 42 are in the BP. In contrast, among the 6251 constitutive core promoters, 1111 are SP, and 1732 are BP. This is because most inducible core promoters have TPM < 1, and constitutive core promoters have a broader range of transcriptional abundance (Fig. 5D). As shown in Figure 5C, core promoters with TPM < 1 have high PSS values or sharp shape. A previous study in Drosophila showed that sharp core promoters are more likely to have restricted tissue-specific expression, whereas broad core promoters tend to have a constitutive temporal expression pattern in Drosophila (Hoskins et al. 2011). Similar to Drosophila, inducible core promoters have restricted condition-specific expression and have a sharper shape than constitutive core promoters, suggesting that the different regulatory mechanisms of transcription initiation between the two classes of core promoters are conserved between yeast and animals.
Strong preference for pyrimidine–purine dinucleotides at yeast TSSs
A strong preference of pyrimidine–purine (PyPu) dinucleotide at TSS, that is a purine at position +1 (TSS), and a pyrimidine at position −1, have been observed in eukaryotes, bacteria, and bacteriophage (Burke and Kadonaga 1997; Hampsey 1998; Zhang and Dietrich 2005; Gleghorn et al. 2011; Zhang et al. 2014). Our data showed that 86.7% of CAGE tags were mapped to PyPu dinucleotide at −1, +1 positions, which is consistent with previous observations based on 5′ SAGE (Zhang and Dietrich 2005). We then investigated whether inducible and constitutive core promoters have different dinucleotide preferences. Using the dominant TSS as the representative TSS for each core promoter, we obtained a consensus sequence surrounding the dominant TSS (±10 nt) for each type of core promoters. As shown in Figure 6A–C, PyPu dinucleotide at position –1,+1 of TSS are highly enriched in both types of core promoters. The most preferred PyPu dinucleotide is CA, following by TA and TG (Fig. 6C). However, constitutive core promoters have a stronger preference for PyPu dinucleotide at TSS than inducible core promoters (95.0% vs. 76.8%) (Fig. 6C). Furthermore, the constitutive core promoters have a much higher frequency of CA dinucleotides than the inducible core promoters (P < 0.01, χ2 test), but inducible core promoters have a stronger preference for G at −1 position (P < 0.01, χ2 test) (Fig. 6C). In addition to the PyPu dinucleotides, the DNA sequences surrounding the dominant TSSs in yeast is enriched of A, especially at position −8 in both types of core promoters, which is a pattern that is not present in other species (Fig. 6A,B). However, the frequency of A at position −8 in constitutive core promoters is higher than inducible core promoters (72.6% vs. 58.8%), supporting different sequence preferences of transcription initiation between the two types of core promoters.
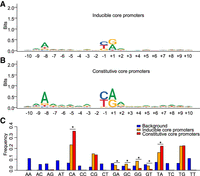
Initiator motif and dinucleotide preference of core promoters in S. cerevisiae. Sequence logo demonstrating a consensus sequence of 20 nt surrounding the dominant TSS of inducible core promoters (A) and constitutive core promoters (B), which likely represents the Initiator element in yeast. (C) TSSs in S. cerevisiae have a strong preference of pyrimidine–purine dinucleotide at [+1,−1] positions in both constitutive and inducible core promoters. The constitutive core promoter has a significantly higher frequency of dinucleotide with “A” and the +1 position (CA, and TA), whereas the inducible core promoter has a significantly higher frequency of “G” at the −1 position. (*) P < 0.01, χ2 test.
Different DNA motifs between inducible and constitutive core promoters
We sought to identify overrepresented DNA motifs in or near the inducible and constitutive core promoters to further explore their different preferences of sequence context. We performed de novo motif discovery for the surrounding sequences of these core promoters (from −100 to +50 nt of TSS). Predicted DNA motifs from each class of core promoters were compared with known binding motifs in S. cerevisiae to identify possible matches (Zhu and Zhang 1999; MacIsaac et al. 2006). Among the top enriched motifs, only two are shared by the two classes of core promoters (Fig. 7). One of them has a consensus sequence of 3′-TATAAA(A)AAA-5 and has significant similarity with the canonical binding sites of TATA-binding protein (TBP), the TATA box (Supplemental Table S6). TBP is a subunit of the TFIID complex in eukaryotes that recruits the transcriptional machinery to the promoter. We found 6843 TATA-box motifs near 1269 inducible core promoters and 1500 constitutive core promoters. The percentage of TATA-box-containing promoters is virtually the same between inducible (24.35%) and constitutive core promoters (24%). A total number of 2284 protein-coding genes were found to be associated with at least one TATA-box motif, which is similar to the number of genes that have zero or one mismatch to the consensus motif of TATA box (TATAWAWR, 676 with 0 mismatches +1781 with 1 mismatch) in a previous study (Rhee and Pugh 2012). In metazoans, transcription typically initiates 25–30 bp downstream from the TATA box. It has been shown that transcription in S. cerevisiae initiates from 60 bp downstream from the TATA box (Rhee and Pugh 2012). Our data showed that the distribution of TATA box forms a sharp peak ∼ 65 bp upstream of TSS, supporting that the preferred locations of TATA box are different between yeast and metazoans. The distributions of TATA box largely overlap between inducible and constitutive core promoters (Supplemental Fig. S10), indicating a similar pattern of distributions and locations of TATA box between the two classes of core promoters.
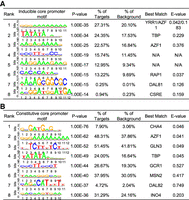
Predicted core promoter motifs in S. cerevisiae. (A) The top enriched motifs present in the inducible core promoter sequences. (B) The top enriched motifs present in the constitutive core promoter sequences. These promoter motifs were predicted by de novo discovery approach for the 150-bp sequence surrounding the dominant TSS (−100 and +50 nt) in each core promoter.
The other shared motif, with a consensus sequence of GGGAAAAAAAA, is present in nearly 50% of core promoters. It is most similar to the binding motif of YRR1 or AZF1 based on motif matches. Both YRR1 and AZF1 are zinc-finger transcription factors. YRR1 is involved in multidrug resistance (Cui et al. 1998), whereas AZF1 is involved in diauxic shift and response to hypoxia (Newcomb et al. 2002). As these transcription factors are only involved in specific cellular processes, we doubt that this motif functions as the binding sites of YRR1 or AZF1, despite their high similarity. We speculated that this motif is the GA element (GAAAA) identified by Seizl et al. (2011). The GA element was found enriched in TATA-less promoters, and it was considered as a functionally substitute for the TATA box (Seizl et al. 2011).
We also noticed that two highly enriched motifs, with consensus sequences of CCCTTTCCCC and AAGGAAAGAAG (Fig. 7A), do not have any significant match with known motif sequences. Because the promoter regions of many eukaryotic genes lack a canonical TATA box, it remains obscure about what motifs are bound by general transcription factors in these genes. We speculated that some of these high-frequency motifs could serve as alternative binding sites for general transcription factors, which play a major role in TSS selection (Pinto et al. 1992; Li et al. 1994). We also inferred the overrepresented motifs associated with the active inducible core promoters in each growth conditions (Supplemental Fig. S11). Although some motifs are enriched in different growth conditions, many appear to be condition-specific, which could be the binding sites of gene-specific transcription factors. Future studies may focus on these motifs to identify their binding proteins and their roles in gene regulation.
Discussion
In this study, we generated a quantitative TSS atlas for an important eukaryotic model organism S. cerevisiae in an unprecedented depth and breadth. Pervasive transcription has been observed in mammalian and yeast genomes (Kapranov et al. 2007; The ENCODE Project Consortium 2007). It was speculated that with the right study, we might observe transcription from the “blank” spaces left in the yeast genome (Libri 2015). We found that in the 12-million bp yeast genome, there are more than 4 million TSS positions supported by at least one CAGE tag, and about 1 million TSS positions were supported by multiple CAGE tags, which is significantly more than any previously identified numbers. However, the biological significance of pervasive transcription is unclear and controversial (Kapranov et al. 2007).
The increase of sequencing depth and examined growth conditions allowed us to identify the TSSs and core promoters for many lowly expressed or condition-specific expressed genes. Based on the nine different TSS maps, we have determined the core promoters for 96% of verified ORFs in S. cerevisiae. In addition to determining the 5′ boundaries for most protein-coding genes in the yeast genome, we also suggested new or alternative translation start codons for 179 ORFs. However, many consensus TSS clusters have not yet been assigned to any known gene features. Transcriptional activities of these clusters are of low abundance in examined conditions. The presence of many low-activity TSS clusters is likely the consequence of the pervasive nature of transcription initiation. However, we cannot exclude the possibility that some could be the functional core promoters of unknown genes. These core promoters can be used as informative markers for identification of novel genes in future studies.
Comparative analysis of quantitative core promoter maps also allowed us to identify two types of core promoters (inducible and constitutive). The constitutive core promoters tend to have higher transcriptional activities than inducible core promoters in all growth conditions examined (Fig. 4C), a more nucleosome-depleted region upstream of TSS (Fig. 4E), a broader promoter shape (Fig. 5D), and stronger preferences of PyPu dinucleotides at TSSs (Fig. 6C). These observations suggest the presence of two distinct regulatory mechanisms of transcription initiation in the unicellular organism.
One of the most interesting findings in this study is widespread core promoter shift and its coupled gene differential expression in response to environmental cues. Alternative promoter usage in different cell types or tissues has been observed in mammals and the fruit fly Drosophila melanogaster (Davuluri et al. 2008; Batut et al. 2013). However, the extent to which the activities of different core promoters in a gene change in response to environmental cues has not been systematically investigated. Our data showed that most yeast genes have multiple core promoters. The activity switch of different core promoters of a gene is prevalent across different growth conditions. Therefore, it appears that alternative core promoter usage is a conserved trait in eukaryotes. We found that most core promoter shifts in yeast are coupled with significant differential gene expression. For microorganisms, modulation of gene expression plays a central role in the adaptation to changing environmental cues (López-Maury et al. 2008). The primary driver of alternative gene expression in response to changing environments is probably the switch of different condition-specific transcription factors, triggered by signal-transduction pathways through sensing extracellular signals (López-Maury et al. 2008). It was found that most of gene differential expression is associated with extensive nucleosome repositioning in the gene promoters (Nocetti and Whitehouse 2016), suggesting that repositioning of nucleosomes in the core promoter regions may also play an important role in the alternative usage of core promoters.
We speculated that the shift of core promoters might serve as a secondary control for further tuning the outcome of gene expression by influencing both transcription and translation processes. These structural differences among core promoters could influence the efficiency of transcription initiation (Kostrewa et al. 2009). In addition, as a direct consequence of core promoter shift, transcripts with various length and sequence of 5′ UTR are generated. Different lengths of 5′ UTR may have different mRNA folding structures, which would change their thermostability. Modulation of mRNA stability is a critical step in the regulation of gene expression. In eukaryotic cells, the decay rates of individual mRNAs vary by more than two orders of magnitude (Harigaya and Parker 2016). Furthermore, the change of 5′ UTR by core promoter shift could theoretically influence translation initiation efficiency, which is the rate at which ribosomes access the 5′ UTR and start translating the ORF (Kudla et al. 2009; Livingstone et al. 2010). Translation initiation efficiency is highly correlated with translation efficiency (Weinberg et al. 2016), and nearly 100-fold range of translation efficiency has been observed in log-phase yeast (Ingolia et al. 2009). Translation initiation is the main rate-limiting steps of gene expression (Pop et al. 2014). Strong secondary structure near the 5′ cap might interfere with binding of the eIF4F-cap-binding complex, and structures within the 5′ UTR can impede the scanning by 40S ribosome, thereby reducing the rate of protein synthesis (Ding et al. 2012). Different 5′ UTR lengths may change the secondary structure near the 5′ cap, which influences translation initiation probabilities (Shah et al. 2013). For instance, insertion of a stem-loop into the 5′ UTR of PGK1 mRNA effectively blocks translation by preventing 40S scanning (Muhlrad et al. 1995). Our previous studies also demonstrated some connections between 5′ UTR lengths and gene expression profiles within and between yeast species (Lin et al. 2010; Lin and Li 2012), suggesting a potentially important functional role of 5′ UTR length in gene regulation. Therefore, more studies would be needed to investigate the functional impacts of core promoter shift and its resulting changes in 5′ UTR length on gene expression, which could potentially uncover a new layer of gene regulatory mechanism.
Methods
Yeast strain and growth conditions
The S. cerevisiae laboratory strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was used as an experimental system to generate condition-specific CTSS maps. The nine growth conditions applied in this study (Table 1) simulate these natural environmental stresses. All incubations were at 30°C, except for when heat stress was applied.
CAGE library preparation and sequencing
Isolation of total RNA was performed with TRIzol (Invitrogen). The total RNA samples were snap-frozen in liquid nitrogen and stored at −80°C. RNA samples were quantified and evaluated for quality and using the Bioanalyzer 2100 (Agilent Technologies). Five micrograms of total RNA was isolated from each sample. Two biological replicates of CAGE libraries were constructed for samples of each growth condition following the nAnT-iCAGE protocol (Murata et al. 2014) by the DNAFORM in Yokohama, Japan. In brief, RNA quality was assessed by Bioanalyzer (Agilent) to ensure that RIN (RNA integrity number) is over 7.0, and A260/280 and A260/230 ratios are over 1.7. First strand cDNAs were transcribed to the 5′ end of capped RNAs, attached to CAGE “barcode” tags. Each nAnT-iCAGE library used linkers with specific barcodes and was sequenced using Illumina NextSeq (single-end, 75-bp reads) at the DNAFORM. The numbers of reads generated from each library were listed in Supplemental Table S1.
CAGE processing, alignment, and rRNA filtering
The sequenced CAGE tags were respectively aligned to the reference genome of S. cerevisiae S288c (R64-2-1) using HISAT2 (Kim et al. 2015). To avoid false TSSs, soft clipping option in HISAT2 was disabled by using “--no-softclip”. The numbers of reads mapped to the S. cerevisiae reference genome are provided in Supplemental Table S2. The reads mapped to rRNA sequences (28S, 18S, 5.8S, and 5S) were identified from read alignments (in SAM format) using rRNAdust (http://fantom.gsc.riken.jp/5/sstar/Protocols:rRNAdust) and were subsequently removed by in-house R scripts (Supplemental Code; R Core Team 2018). Tags mapped to multiple genomic regions (SAM MAPQ < 20) were excluded for further analysis. The unique 5′ ends of tags were identified as CAGE tag–defined TSSs (CTSSs) by in-house R scripts (Supplemental Code). The replicates of CAGE tags obtained from the same growth condition were merged. The numbers of CAGE tags supporting each CTSS were counted and normalized to tag per million (TPM) using the CAGEr package (Haberle et al. 2015) in R Bioconductor.
Analysis of mapped CAGE tags
CTSSs with a minimum TPM value of 0.1 were used as input for tag clustering to infer putative core promoters. CTSSs separated by <20 bp were clustered into a larger transcriptional unit, called tag cluster (TC). Only TCs with a minimum of 0.2 TPM were used for further analysis. For each TC, we calculated a cumulative distribution of the CAGE tags to determine the positions of the 10th and 90th percentile, which were considered as its boundaries. TCs were first generated from each sample separately. Based on TC locations across nine samples, if two TCs are located within <50 bp, they likely belong to the same core promoters, so they were aggregated into a consensus cluster. Gene Ontology (GO) term enrichment analysis was carried out by Go-TermFinder (https://go.princeton.edu/cgi-bin/GOTermFinder). Redundant GO terms (similarity > 0.7) were removed and the scatterplot of GO terms in a two-dimensional space was generated by using REVIGO (Supek et al. 2011).
Experimental validation
We modified the CAGE protocol (Murata et al. 2014) to experimentally validate the 5′ end of transcripts for selected genes. Specifically, total RNA was isolated with TRIzol (Invitrogen) from yeast cells grown in each selected condition. Five micrograms of mRNA from each sample was reversed transcribed using gene-specific primers (mixing 100 µM CIK1-R, FIG2-R, and ICL1-R primers before use). Biotin was ligated to the cap structure at the 5′ end after diol structure was oxidized with NaIO4. To cleavage single-stranded RNA regions, RNase I was used to treat the samples and biotinylated caps were captured by M-270 streptavidin beads. After 5′ complete cDNA was released from magnetic beads, 5′ linkers were ligated to the single-stranded cDNA. A first round of PCR was performed to amplify targeted fragments. Nested PCR with nested primers was performed to improve amplification specificity. All primers and linker sequences used in this study were provided in Supplemental Table S6.
Core promoter shift and gene differential expression analyses
The degree of core promoter shift was calculated by using DS = log2[(Pt/Dt)/(Pc/Dc)]. Pt and Dt are the transcript abundance (TPM) of the proximal and distal core promoters in the treatment condition, and Pc and Dc are the transcript abundance (TPM) of the proximal and distal core promoters in the control (YPD). DS = 0 means no core promoter shift. DS > 0 means shift toward the proximal core promoter in the treatment, and DS < 0 means a shift toward the distal core promoter. We implemented the χ2 test to infer its statistical significance. We identified differentially expressed genes in all eight comparisons using DESeq2 (Love et al. 2014). In both promoter shift and differential gene expression analyses, P-values of χ2 tests were adjusted with the Benjamini–Hochberg method (Benjamini and Hochberg 1995) to account for the multiple comparisons issue. Significant core promoter shift and differentially expressed gene was defined if adjusted P-value (FDR) < 0.05.
Promoter shape score
We calculated promoter shape score (PSS) to quantify the shape of a core promoter based on the distribution of CAGE tags within
a core promoter and promoter width. The PSS was calculated using the following equation:
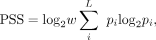 where p is the probability of observing a CTSS at base position i within a core promoter; L is the set of base positions that have normalized TSS density ≥ 0.1 TPM; and w is the promoter width, which was defined as the distance (in base pairs) between the 10th and 90th quantiles. This width
marks the central part of the cluster that contains ≥80% of the CAGE signal.
where p is the probability of observing a CTSS at base position i within a core promoter; L is the set of base positions that have normalized TSS density ≥ 0.1 TPM; and w is the promoter width, which was defined as the distance (in base pairs) between the 10th and 90th quantiles. This width
marks the central part of the cluster that contains ≥80% of the CAGE signal.
Sequence context analyses and de novo promoter motif discovery
Dinucleotide frequencies were calculated with sequences extracted with BEDTools nuc from [−1,+1] of TSS in the S. cerevisiae genome (Quinlan 2014). We performed de novo motif discovery with HOMER (http://homer.ucsd.edu/homer/). The sequences were retrieved from −100, +50 nt of the dominant TSS from each core promoter. The predicted motifs were compared with known motifs of transcription factors obtained from MacIsaac et al. (2006) and Saccharomyces cerevisiae Promoter Database (SCPD) (Zhu and Zhang 1999) using Tomtom module (Gupta et al. 2007) of the MEME Suite (Bailey et al. 2015).
Data access
The raw CAGE sequencing data generated in this study have been submitted to the NCBI BioProject database (BioProject; https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA483730. The quantitative maps of TSS and core promoters generated in this study can be visualized and downloaded from the YeasTSS database (http://www.yeastss.org) (McMillan et al. 2019).
Acknowledgments
This study was supported by the start-up fund and Beaumont Award from Saint Louis University to Z.L. We thank Dr. Craig Kaplan, Dr. Dapeng Zhang, Dr. Yong Xue, Dr. Hong Qin, and three anonymous reviewers for their constructive comments, which have significantly improved this manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245456.118.
- Received October 22, 2018.
- Accepted May 7, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








