
Beta-binomial modeling of CRISPR pooled screen data identifies target genes with greater sensitivity and fewer false negatives
- Hyun-Hwan Jeong1,2,
- Seon Young Kim1,2,
- Maxime W.C. Rousseaux1,2,5,
- Huda Y. Zoghbi1,2,3,4 and
- Zhandong Liu2,3
- 1Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 2Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital, Houston, Texas 77030, USA;
- 3Department of Pediatrics, Baylor College of Medicine, Houston, Texas, USA;
- 4Howard Hughes Medical Institute, Houston, Texas 77030, USA
Abstract
The simplicity and cost-effectiveness of CRISPR technology have made high-throughput pooled screening approaches accessible to virtually any laboratory. Analyzing the large sequencing data derived from these studies, however, still demands considerable bioinformatics expertise. Various methods have been developed to lessen this requirement, but there are still three tasks for accurate CRISPR screen analysis that involve bioinformatic know-how, if not prowess: designing a proper statistical hypothesis test for robust target identification, developing an accurate mapping algorithm to quantify sgRNA levels, and minimizing the parameters that need to be fine-tuned. To make CRISPR screen analysis more reliable as well as more readily accessible, we have developed a new algorithm, called CRISPRBetaBinomial or CB2. Based on the beta-binomial distribution, which is better suited to sgRNA data, CB2 outperforms the eight most commonly used methods (HiTSelect, MAGeCK, PBNPA, PinAPL-Py, RIGER, RSA, ScreenBEAM, and sgRSEA) in both accurately quantifying sgRNAs and identifying target genes, with greater sensitivity and a much lower false discovery rate. It also accommodates staggered sgRNA sequences. In conjunction with CRISPRcloud, CB2 brings CRISPR screen analysis within reach for a wider community of researchers.
Genetic screens have become a favored tool for gathering information about disease pathogenesis and cellular biology. Initially, these screens were performed using chemical mutagenesis or RNA inference (RNAi), which are effective but laborious processes (Schlabach et al. 2008; Silva et al. 2008; Park et al. 2013; Mohr et al. 2014; Simon et al. 2015). Larger-scale, pooled approaches were finally made feasible by the advent of microarray (Luo et al. 2009; Gilbert et al. 2014; Shalem et al. 2014; Wang et al. 2014; DeJesus et al. 2016). Pooled shRNA (short-hairpin RNA) libraries can be barcoded and packaged into viruses, which are used to infect a population of cells that are then selected for a desired phenotype (e.g., growth or fluorescence). Hybridizing microarray soon followed for hit identification (Paddison et al. 2004). In later iterations of this approach, next-generation sequencing (NGS) was used to identify hits (Hu and Luo 2012).
The development and optimization of clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR/Cas9) systems have propelled pooled screen approaches into even wider use. Besides the relative simplicity and low cost, the robustness of hit identification has reduced the requirement for redundancy in the number of targeting single-guide RNAs (sgRNAs), which allows the same size library to be more diverse (Sanjana et al. 2014; Xu et al. 2015). Moreover, these pooled libraries have been made accessible through repositories such as Addgene (https://www.addgene.org/). CRISPR/Cas9 pooled screens are thus within the technical reach of most biomedical researchers (Doench 2017). For all this accessibility on the experimental side, however, the resulting bioinformatics data sets are enormous and complex. The analysis of thousands of genetic perturbations demands considerable bioinformatics expertise.
In our experience, most users are unaware of whether their tool of choice models the data according to Poisson, negative binomial, or Gaussian distribution, or of the relative strengths and weaknesses of these models. The current roster of tools bears the imprint of the history of RNA-seq data analysis: RNA-seq data were initially modeled using Poisson distributions (Marioni et al. 2008), which is a natural choice for simple read counts. Poisson distribution assumes that the mean and variance are equal, however, and with biological data, the variance is often greater than the mean. Analytic methods therefore turned to negative binomial distribution, which can handle overdispersed data (Anders and Huber 2010; Love et al. 2014). A number of popular tools still use negative binomial distribution to analyze sgRNA screen data (Li et al. 2014; Spahn et al. 2017), even though the structure of the sgRNA screen data is very different from that of RNA-seq. In the latter, there is huge variation in transcript lengths, from 60 bp to 2.4 Mbp, but all sgRNAs for any given gene are designed to have the same length. This often leads to the variance being less than the mean (Supplemental Fig. S1). We hypothesized that a beta-binomial model, in which the variance can be either larger or smaller than the mean, would better fit the data and more accurately identify changes in sgRNA.
We therefore developed CB2, a new web-based algorithm that uses beta-binomial distribution, and compared its performance with that of the eight most commonly used algorithms (HiTSelect [Diaz et al. 2015], MAGeCK [Li et al. 2014], PBNPA [Jia et al. 2017], PinAPL-Py [Spahn et al. 2017], RIGER [Luo et al. 2009], RSA [König et al. 2007], ScreenBEAM [Yu et al. 2016], and sgRSEA [https://cran.r-project.org/web/packages/sgRSEA/]), which encompass both parametric and nonparametric approaches (Table 1). We applied all these methods to 10 different biological data sets, taken from fields ranging from cancer to basic cell biology (Koike-Yusa et al. 2014; Parnas et al. 2015; Evers et al. 2016; Golden et al. 2017; Li et al. 2018; Sanson et al. 2018).
Statistical models used by CB2 and existing methods
Results
CB2 is more sensitive in target gene identification than existing methods
Identifying candidates by a statistical hypothesis test is a key component in any screen analysis. In CB2, we adapted a beta-binomial model (Baggerly et al. 2003) with a modified Student's t-test to measure differences in sgRNA levels, followed by Fisher's combined probability test (Fisher 1925) to estimate the gene-level significance. We chose Fisher's method for two reasons: first, to keep the entire pipeline parametric and, second, to keep CB2 as fast as possible (robust rank aggregation [RRA] requires permuting the data, a nonparametric feature, so it runs slower than Fisher's method). Furthermore, when we compared Fisher's method against RRA, we found that RRA is not effective in combining the P-values estimated by beta-binomial distribution (Supplemental Fig. S2).
We compared CB2 with eight state-of-the-art methods on three benchmark data sets evaluating gene essentiality (Evers et al. 2016) using different technologies: CRISPR nuclease gene knockout via Cas9 (CRISPRn) and CRISPRinterference (CRISPRi; a CRISPR/Cas9 system with a catalytically inactive Cas9 fused to the transcriptional repressor KRAB, which results in gene repression). These benchmark data sets (CRISPRn-RT112, CRISPRn-UMUC3, and CRISPRi-RT112) were constructed based on 46 genes that are essential for cell survival and 47 genes that are nonessential. We first tested whether these methods could easily distinguish between essential and nonessential genes. We found that each method clearly discriminates essentiality by their gene rankings (Supplemental Fig. S3A). In addition, gene rankings obtained from each method, except PBNPA for the CRISPRi-RT112 data set, are highly correlated (R2 is [0.86, 0.98] for CRISPRn-RT112, [0.85, 0.98] for CRISPRn-UMUC3, and [0.72, 0.96] for CRISPRi-RT112) (Supplemental Fig. S3B). CB2, ScreenBEAM, and MAGeCK produced very similar gene rankings across all the benchmark data sets. We also compared the precision-recall (PR) and receiver operating characteristic (ROC) curves across the methods and calculated the area under the curve (AUC) of each. CB2 recorded the best PR-AUC and ROC-AUC scores for both CRISPRn screen data sets (Supplemental Figs. S4, S5). HiTSelect had the best PR-AUC and ROC-AUC scores for the CRISPRi-RT112 data set, for which CB2 achieved comparable scores (Supplemental Fig. S6). Although the gene ranking is similar among these methods (Supplemental Fig. S3), the estimated P-values and false-discovery rates (FDRs) are very different. These results highlight the importance of using FDR to guide the gene selection process.
Although several CRISPR screens (Zhou et al. 2014; Parnas et al. 2015; Aguirre et al. 2016) have prioritized candidate hits by ranking, they do not provide statistical estimates of error rates. These methods, therefore, rely on an arbitrary rule to select the top candidate genes and are prone to biased selections and high hit attrition rates. One solution is candidate selection by a quantitative statistical measure such as a P-value or a false-positive rate (FDR) cut-off. To assess the detection powers of FDR of established CRISPR screen analysis methods and CB2, we measured the F1-score (2 × precision × recall/precision + recall) of each method, namely, the harmonic average of the precision and the recall, for FDR thresholds ranging from 10% to 0.01%. CB2 outperformed all other methods at every FDR cut-off level, and all other methods lost their detection powers at more rigorous FDRs (Fig. 1A,B; Supplemental Fig. S7). In other words, all methods showed a small type-I error owing to the strong lethality phenotype of the CRISPR assay, but CB2 showed a significantly lower type-II error than the other methods (Supplemental Fig. S8). Across all paradigms tested with different FDR cut-offs, CB2 performed the best, with a much larger F1-score and recall. Thus, CB2 is both sensitive and specific in selecting candidate genes.
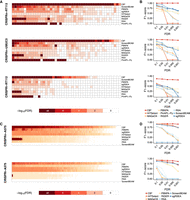
CB2 offers robust target identification with high precision and recall. (A,B) Benchmark results using data from Evers et al. (2016). (A) Heatmaps illustrate FDRs of gene statistics from each of nine leading high-complexity pooled screen analysis tools. (B) F1-score measurements at different FDR cut-offs across all methods. At commonly used FDR cut-offs, CB2 can identify most of the essential genes with high rates of precision and recall. (C,D) Same representation as in A and B, using data from Sanson et al. (2018).
To understand the differences produced by these methods, we next tested a prototypical essential gene, RPL5, to compare the gene-level enrichment across data sets and analytical tools. In the first CRISPR screen on an RT112 cell line, we expected to see the depletion of sgRNAs targeting RPL5 in group T1. Out of the 10 sgRNAs that target this gene in the CRISPRn-RT112 data set, six showed a strong decrease in the group T1. CB2 estimated an FDR of 2.07 × 10−19, whereas only three other methods (HiTSelect, ScreenBEAM, and sgRSEA) estimated FDR to be <0.01 (Fig. 2A). Next, we looked at the same gene in the UMUC3 cell line. Five of the 10 sgRNAs targeting RPL5 decreased in group T1, and all five sgRNAs were among those identified in RT112 cell line. CB2 estimated an FDR of 3.81 × 10−10 for RPL5, whereas none of the other methods identified it to be significantly depleted with an FDR cutoff below 0.01 (Fig. 2B). Lastly, in the CRISPRi-RT112 data set, three of seven sgRNAs indicated depletions, but only CB2 was able to estimate the FDR of 2.78 × 10−8, and the other methods did not count RPL5 as a hit in the data set (Fig. 2C). Overall, CB2 produced more reliable hit identification than other methods based on statistical cut-offs for the gene tested (COPS8 and RPL27) (Supplemental Figs. S12, S13).
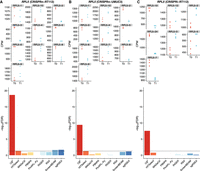
CB2 detects essential genes missed by other leading methods: the case of RPL5. sgRNA quantification for RPL5 in cell line (A) RT112, (B) UMUC3 using CRISPRn, and (C) RT112 using the CRISPRi library. The top panels show counts per million (CPM) of sgRNAs that target RPL5 for each group (T0 and T1), and the bottom panels indicate the reported the FDR for RPL5 in each screen across all the methods. A horizontal line at FDR = 0.01 is used as a threshold for statistical cutoff. CB2 outperforms all other methods of identifying RPL5 as an essential gene across all benchmark data sets.
We performed the same analysis on two distinct data sets (Sanson et al. 2018) to determine how CB2 performs compared with other methods for genome-wide screening analysis. Sanson et al. (2018) used novel optimized libraries for genome-wide CRISPRn (Brunello), CRISPRi (Dolcetto), and CRISPRa (Calabrese) screening and showed these libraries outperform other previously established libraries, such as GeCKO (Sanjana et al. 2014) and hCRISPRi-v2 (Horlbeck et al. 2016). For our analysis, we chose two screens for benchmarking, both data sets from a dropout screen in A375 melanoma cells: One used the Brunello CRISPRn library with tracr-v2 tracrRNA (CRISPRn-A375); the other used the Dolcetto CRISPRi Set A library (CRISPRi-A375). Each data set contains a control sample (plasmid DNA) and three biological replicates. We used the gold-standard gene sets of 1580 essential and 927 nonessential genes reported by Hart et al. (2014, 2015) to assess the performance of the methods. (We excluded PinAPL-Py from benchmarking because it does not report statistics when the input contains only one control sample.) CB2 outperformed other methods at the stringent FDR cutoff level (Fig. 2; Supplemental Fig. S9). CB2 outperformed all other methods in F-1 and PR measures at the stringent FDR cut-offs on A375 genome-wide screen data sets of Sanson et al. (2018). F1-score (top), precision (middle), and recall (bottom) for each method on two benchmark data sets are presented as a function of FDR cut-off values (Fig. 1C,D; Supplemental Fig. S9) and provided higher AUC values of PR and ROC curves than the other methods (Supplemental Figs. S10, S11).
These results indicate that CB2 more accurately estimates the gene-level FDR. The use of FDR in selecting hits is critical in real data analysis because the arbitrary selection of top genes is purely heuristic. CB2 is better at identifying true hits based on multiple concordant sgRNAs targeting the same gene.
CB2 is more specific in target gene detection than existing methods
To test the idea that a beta-binomial model would better fit the data and more accurately identify changes in sgRNA, we compared the sgRNA level statistics on several CRISPR pooled libraries containing nontargeting sgRNAs as negative controls. Nontargeting sgRNAs are not supposed to show any differential enrichment and can be used to assess the quality of the method. Parnas et al. (2015) used the Mouse CRISPR Knockout Pooled Library for their genome-wide screen (GeCKO v2; Addgene 1000000052, 1000000053), which contains 1000 nontargeting sgRNAs. We therefore used this data set to measure the specificity with which CB2 and MAGeCK detect true negatives. We compared the unadjusted P-values for sgRNAs because the FDR is controlled at the gene level.
CB2 showed greater specificity (the proportion of actual negatives that are correctly identified) than MAGeCK across a wide range of P-value thresholds. At a P-value threshold of 0.01, CB2 had a specificity of 86% versus MAGeCK's 68% (Fig. 3A). Next, we plotted the log-fold change against the P-value levels in a volcano plot. The majority of the negative control sgRNAs were correctly detected by CB2, whereas MAGeCK showed a one-side long tail for positively changed sgRNAs, producing inflated P-values for a group of negative controls (Fig. 3B). Many of these false positives showed extremely low P-values (ranging from 10−5 to 10−40), indicating a strong selection bias. To understand the impact of this selection bias, we analyzed the rest of the sgRNA library with both methods. At the same threshold (P < 0.01), CB2 selected 12,648 sgRNAs, whereas MAGeCK selected 31,381 sgRNAs; 2971 sgRNAs were identified by both methods (Fig. 4B). We applied the same analysis to the CRISPRn-A375 data set (Sanson et al. 2018), which contains 1000 nontargeting sgRNAs. CB2 shows higher specificity than MAGeCK, except when setting a P-value cut-off at 0.2. Similar P-value distributions shown in Figure 3 were also found for this data set (Supplemental Fig. S14).

Comparison of false-positive rate for nontargeting sgRNAs on screen data of Parnas et al. (2015). (A) Specificity comparison between CB2 and MAGeCK for the six different P-value thresholds. The y-axis indicates specificity, and the x-axis indicates the level of the P-value threshold for the specificity calculation. (B) Volcano plots of the P-value of nontargeting sgRNAs. The y-axis indicates the negative logarithm value of P-value, and the x-axis indicates the log2 value of fold-change. All of the data points are from negative control sgRNAs. False positives were plotted in red. Horizontal blue lines at P = 0.01 indicate the threshold for statistical cutoff.
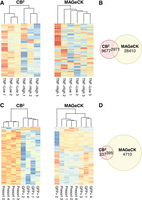
Discrepancy of sgRNA-level statistics between CB2 and MAGeCK in two public CRISPR pooled screens. (A) Heatmaps of normalized read counts of the detected sgRNAs from the screen data of Parnas et al. (2015). (Left) A heatmap of sgRNAs detected by CB2 only. (Right) A heatmap of sgRNAs detected by MAGeCK only. (B) Venn diagrams of sgRNAs detected by CB2 and MAGeCK from the screen data of Parnas et al. (2015). (C,D) Same representations of A and B using data from Li et al. (2018).
We plotted sgRNAs unique to each method on a heatmap, which showed a high concordance within each experimental group for sgRNAs unique to CB2 (Fig. 4A). In contrast, sgRNAs identified by MAGeCK showed a much noisier pattern, and samples from the same experimental group could not be clustered together based on these differentially enriched sgRNAs (Fig. 4A).
Next, we performed the same sgRNA-level comparisons on two additional data sets. In the first study, a differentiation screen was conducted to identify target genes that maintain naive pluripotency (Li et al. 2018) using the Mouse Improved Genome-wide Knockout CRISPR Library v2 (Addgene 67988). The library contains 91,319 sgRNAs targeting 18,542 mouse genes. To identify differentially enriched sgRNAs, we kept the same threshold (P < 0.01) for both CB2 and MAGeCK. CB2 identified 732 sgRNAs whereas MAGeCK identified 5105 sgRNAs (395 sgRNAs shared between the two) (Fig. 4C,D), and we observed the same trend as in screening data set of Parnas et al. (2015) screening data set (Fig. 4A,B). We found the same trend on the data sets of Evers et al. (2016) (Supplemental Fig. S15). Thus, CB2’s accurate sgRNA-level statistics are attributable to its use of the beta-binomial model.
CB2 provides more accurate alignment without parameter tuning
Many CRISPR pooled screens use in-house scripts to quantify sgRNA abundance (Gilbert et al. 2014; Golden et al. 2017; Iorio et al. 2018; Li et al. 2018) or other alignment algorithms for RNA-seq (Sanjana et al. 2014; Hart et al. 2015; Parnas et al. 2015; DeJesus et al. 2016). These codes are often not shared publicly and are not easily reusable. Both MAGeCK and PinAPL-Py provide an integrated mapping function, but PinAPL-Py requires complex parameter tuning and MAGeCK samples only the first million reads to estimate the location of sgRNAs in FASTQ files. Furthermore, there is no systematic comparison of mapping accuracy in the literature, and users lack reliable guidelines for selecting mapping tools. We therefore introduced an adaptive hash-mapping algorithm into CB2 and tested all three methods on six published data sets (Supplemental Table S1).
CB2 showed consistently greater mapping accuracy than MAGeCK and PinAPL-Py (Fig. 5A). To understand why, we studied the reads that are mapped by CB2 but not MAGeCK or PinAPL-Py in the CRISPRn-RT112 data set (Evers et al. 2016). MAGeCK mapped 64% of the reads compared with 75% by CB2 (Fig. 5A). This is primarily because MAGeCK estimates the trimming windows using the first N reads from the input (N is 100,000 by default). There is no guarantee that these windows are optimal for the rest of the input files. If a sgRNA locates outside of the precomputed windows, MAGeCK will fail to detect it (Fig. 5B). PinAPL-Py does not precompute sgRNA locations based on a subset of reads but uses Cutadapt (Martin 2011) for flexible trimming followed by the Bowtie 2–based alignment (Langmead and Salzberg 2012). We found that PinAPL-Py failed to identify some of the sgRNAs because reads fail to align owing to the incorrect trimming from Cutadapt even under several different tuning parameters (Fig. 5B). This is likely because of the frequent indels that occurred in the 5′ adapter sequence region of the reads (see Fig. 5B): Usually the quad-nucleotide sequence “CACC,” which is part of the U6 promoter, precedes the sgRNA sequence. In contrast, the “GTTT” sequence, which is the first 4 nucleotides (nt) of the sgRNA scaffold sequence, was present in all the reads. Given the fidelity of the GTTT sequence, the sequences mapped by CB2 but missed by other algorithms are likely accurate and not false positives. CB2 is currently the only CRISPR/Cas9 online screen analysis tool with parameter-free mapping and high accuracy in sgRNA quantification.

CB2 outperforms MAGeCK and PinAPL-Py in the percentage of mapped reads over six benchmark data sets. (A) Read mappability of CB2, MAGeCK, and PinAPL-Py across six different data sets. (B) Representative examples of reads that were not mapped by MAGeCK or PinAPL-Py. Adapters are highlighted with green; sgRNAs, with red. Yellow boxes show the predicted locations of sgRNAs by each method. Several parameters were used to optimize performances of PinAPL-Py.
CB2 is accessible, secure, and easy to learn with CRISPRcloud
We had previously developed a web-based application called CRISPRcloud that could run any statistical testing and mapping algorithm through the cloud-based infrastructure provided by Amazon Web Service (AWS) (Jeong et al. 2017). We implemented CB2 in the platform and added new features to increase speed and data security (Supplemental Fig. S16). CRISPRcloud is compatible with most modern web browsers (Google Chrome version 69 and later, Apple Safari version 11 and later, and Mozilla Firefox 48.0 and later) and operating systems (iOS, Windows, and Linux). Our fast client-side sgRNA mapping program reduces input files of several gigabytes into a single megabyte-sized file. By transferring a much smaller file through the Internet, CRISPRcloud decreases transfer time and prevents the sharing of raw input files, thereby eliminating downloading errors and data privacy issues in one step. Our adaptive mapping algorithm, via Angular (https://angular.io/) and TypeScript (https://www.typescriptlang.org), provides an open-source front-end web application platform. The enormous computing power needed to perform these operations mean that platforms built with a centralized server solution will have load-balancing problems when many users submit their requests simultaneously, leading to the longer user waiting times and raising the risk of system-wide failure. CB2 therefore provides a decentralized, cloud-computing-based, scalable service through a combination of AWS infrastructure that includes Amazon Elastic Compute Cloud (EC2) (https://aws.amazon.com/ec2/), Amazon Simple Storage Service (S3) (https://aws.amazon.com/s3/), and Amazon Simple Queue Service (SQS) (https://aws.amazon.com/sqs/). With this infrastructure, we launched a web service of CRISPRcloud. CRISPRcloud enables researchers with no programming background to preprocess, check the quality, statistically analyze, query, and visualize their CRISPR/Cas9 pooled screening data (Supplemental Table S2).
Discussion
The number of data sets for CRISPR/Cas9 screens in NCBI Gene Expression Omnibus have more than tripled each of the past 3 yr (39 data sets in 2015, 121 data sets in 2016, and 408 data sets in 2017). This expansion has outpaced the development of methods for analyzing the data, most of which use statistical models that are better suited to RNA-seq than to sgRNA data. Here we took into account the difference between the two types of data to develop a new algorithm, CB2, and show that it is more sensitive, specific, and selective than eight other leading tools.
We focused first on the central task for any analytic tool being applied to sgRNA data: statistical hypothesis testing to identify target genes accurately from the screening data. Several methods that facilitate analysis of RNAi pooled screening data (König et al. 2007; Luo et al. 2008; Shao et al. 2013; Dutta et al. 2016) are not compatible with CRISPR/Cas9 pooled screening data because of differences in effect size, sequence determinants, and on- versus off-target effects (Li et al. 2014). MAGeCK was the first tool specifically developed to analyze CRISPR/Cas9 pooled screening data, and it combines a negative-binomial distribution model with a modified robust ranking aggregation (α-RRA) algorithm (Li et al. 2014). Subsequent methods (Table 1) used different strategies to improve the accuracy of data analysis, but to our knowledge, there has never been a thoroughgoing attempt to benchmark these methods and determine which performs best with sgRNA data. Our choice of the beta-binomial distribution, which is not the approach used by any of these analytic tools, was justified by both the theoretical and empirical considerations and proved able to provide far fewer false positives than these other methods at comparable FDR thresholds.
CB2 also addressed another difficult task: quantifying sgRNA from NGS data. Except for the quantification algorithm provided by MAGeCK, most studies use in-house algorithms or extend established methods that were optimized for RNA-seq. CB2 proved capable of fast and accurate alignment, with the ability to handle indels.
Last but not least is the challenge of making powerful tools readily accessible to the research community. Of the existing tools, PinAPL-Py (Spahn et al. 2017) and CRISPRcloud (Jeong et al. 2017) are the only two that support a graphical web interface and require no additional program installation. These programs are an important first step toward enabling the scientists who are actually generating the CRISPR/Cas9 screen data to analyze their large data sets. They still have limitations however: For PinAPL-Py, users still need to provide the adapter sequence to be trimmed, the error tolerance rate, and the quality threshold for trimmed reads. CRISPRcloud is the only framework into which the user can plug in any statistical tools or mapping algorithms, but transferring a large amount of sequence data over the Internet has inherent disadvantages such as long transfer times, vulnerability to file copying errors, and possible data security breaches. By taking advantage of the CRISPRcloud framework, CB2 is fully web-based and designed to require only the minimal number of parameters for data analysis, because fewer parameters mean shorter learning curves for the majority of users. CB2’s power and accessibility will enable more laboratories to extract biologically relevant discoveries from CRISPR pooled screens.
Methods
Statistical hypothesis testing using beta-binomial distribution for sgRNA-level differential analysis
We adapted a beta-binomial model proposed for serial analysis of gene expression (SAGE) by Baggerly et al. (2003). Specifically, let pi be the true proportion of an sgRNA in sample i. We assume the value of pi can vary from sample to sample and follows a beta distribution, pi ∼ Beta(α, β). Let Xi denote the number of read counts for a sgRNA in the ith sample. We assume Xi follows a binomial distribution, Xi|pi ∼ Binomial(ni, pi), where ni is the total number of mapped reads in sample i. To combine the estimated 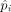 across multiple samples of the same treatment group, we proposed a linear model
across multiple samples of the same treatment group, we proposed a linear model 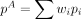 , where i is the index for samples and w is the weight vector for samples in group A. Baggerly et al. (2003) proved that as long as wT1 = 1, the expectation of E(pA) is unbiased. The value of w is estimated through gradient descent methods by minimizing the variance on
, where i is the index for samples and w is the weight vector for samples in group A. Baggerly et al. (2003) proved that as long as wT1 = 1, the expectation of E(pA) is unbiased. The value of w is estimated through gradient descent methods by minimizing the variance on 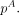 Baggerly et al. (2003) showed that
Baggerly et al. (2003) showed that 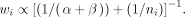
CB2 performs the sgRNA-level differential analysis between two groups using a Student's t-test–like statistic (Baggerly et al. 2003):
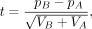 where pA and pB are the proportions of sgRNA, and VA and VB are the group variances of sgRNA, for groups A and B, respectively. Test statistic t represents the strength of the difference of sgRNA abundance between groups A and B. In other words, a large positive t-value indicates that the quantity of sgRNA in group B is greater than in group A, and a large negative t-value indicates that the quantity of sgRNA in group B is less than that in group A.
where pA and pB are the proportions of sgRNA, and VA and VB are the group variances of sgRNA, for groups A and B, respectively. Test statistic t represents the strength of the difference of sgRNA abundance between groups A and B. In other words, a large positive t-value indicates that the quantity of sgRNA in group B is greater than in group A, and a large negative t-value indicates that the quantity of sgRNA in group B is less than that in group A.
The variance is estimated by
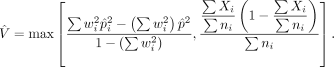
To measure the statistical significance of the difference, we approximate the p-value of a given t in a Student's t-distribution with a degree of freedom (df) defined by
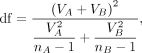 where nA and nB are the numbers of replicates in groups A and B.
where nA and nB are the numbers of replicates in groups A and B.
sgRNA p-value aggregation for gene-level statistics
Because multiple significant sgRNAs targeting the same gene hold greater biological significance than a single significant
sgRNA, we must aggregate p-values to increase confidence in target identification. To do so, we combine p-values of sgRNAs for a target gene using Fisher's method (Fisher 1925) to assess overall differences at the gene level. The combined chi-square statistical test is used:
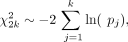 where k is the number of sgRNAs targeting a gene in the screen, and pj is the p-value of jth sgRNA for the gene. χ2 follows a chi-squared distribution with 2k degrees of freedom. To correct for multiple hypothesis testing, we adapted the Benjamini–Hochberg procedure (Benjamini and Hochberg 1995) to estimate the FDR.
where k is the number of sgRNAs targeting a gene in the screen, and pj is the p-value of jth sgRNA for the gene. χ2 follows a chi-squared distribution with 2k degrees of freedom. To correct for multiple hypothesis testing, we adapted the Benjamini–Hochberg procedure (Benjamini and Hochberg 1995) to estimate the FDR.
Gene-level statistics benchmarking on existing methods
We used three different CRISPRn/CRISPRi pooled screen data sets (RT112 and UMUC3 cell line screens with CRISPR; RT112 cell line screen with CRISPRi) (Evers et al. 2016) and two different genome-wide CRISPRn/CRISPRi pooled screen data sets (A375 cell line screens) (Sanson et al. 2018), which provide ground-truth labels of essentiality for each gene. With those screening data sets and labels, we benchmarked the accuracy of essential gene detection by CB2 with eight other published methods (HiTSelect, MAGeCK, PBNPA, PinAPL-Py, RIGER, RSA, ScreenBEAM, and sgRSEA). We computed the FDR for each gene from each method in the benchmark and set five different levels of FDR cut-off (0.1, 0.05, 0.01, 0.005, 0.001) for essential gene classification. For example, if we set FDR cut-off to 0.1, then a gene is predicted to be essential in the cell line if the FDR of the gene falls below the cut-off value. We calculated recall (a recall value close to one indicates a prediction with a low false-negative rate), precision (a precision value close to one indicates a prediction with a low false-positive rate), and F-measure (the harmonic mean of PR) of all the methods at each FDR level. To allow each method to archive its best performance, we tuned parameters for the five parameter-tunable methods: MAGeCK (permutation parameter for RRA test), PBNPA (no.sim parameter), RIGER (alpha parameter), sgRSEA (multiplier parameter), and ScreenBEAM (burnin parameter) on CRISPRn-RT112 data set. The F-measure was used as a measure for the parameter tuning. Most of the methods showed robust performance regardless of the varied parameters, except sgRSEA and RIGER (Supplemental Fig. S17). Therefore, we used the default parameter for MAGeCK, PBNPA, and ScreenBEAM for other data sets and used an optimized parameter for sgRSEA and RIGER. We also calculated the AUC of the PR curves and ROC curves of all the methods with FDR values.
CB2
The CB2 R package was used in the benchmarking (R Core Team 2019). Benchmarking of CB2 was performed without parameter tuning because CB2 is parameter free. FDR values for negative changes between two different groups from CB2 statistical analysis were used for benchmarking.
HiTSelect
We ran the HiTSelect MATLAB package (https://github.com/diazlab/HiTSelect). Normalization by sequencing depth option was selected for benchmarking.
MAGeCK
MAGeCK version 0.5.8 was used for benchmarking. We ran MAGeCK with the “mageck test” command with the following parameters: “--norm-method,” “median,” and “--adjust-method” “fdr.” We performed 100 permutations for the modified robust ranking aggregation (α-RRA) algorithm to estimate the gene-level statistics on the benchmark data sets.
ScreenBEAM
The ScreenBEAM R package (version 1.0.0, https://github.com/jyyu/ScreenBEAM) was used for benchmarking “data.type” parameter was set as “NGS,” and “do.normalization” was set as TRUE, and “nitt” and “burnin” parameters for Bayesian computing were set at 15,000 and 5000. ScreenBEAM does not provide the one-sided p-value for negative selection, so for the FDR comparison with other methods, we changed the FDR of a gene to one if the β of the gene is greater than zero.
PinAPL-Py
We used the PinAPL-Py website (http://pinapl-py.ucsd.edu) to perform the benchmarking. For the sgRNA read counting, we used “GGCTTTATATATCTTGTGGAAAGGACGAAACACCG, GCTTTATATATCTTGTGGAAAGGACGAAACACCG,” and “CTTTATATATCTTGTGGAAAGGACGAAACACCG” for “seq_5_end” parameters of “CRISPRn-RT112,” “CRISPRn-UMUC3,” and “CRISPRi-RT112” data sets. We used CPM normalization and set the GeneMetric parameter as “aRRA” to perform a modified robust ranking aggregation (α-RRA). We used the combined FDR values for each gene in the benchmarking.
RIGER
We used the Java implementation of RIGER (version 2.0; https://github.com/broadinstitute/rigerj) to perform the benchmarking. We set the “alpha” parameter at 0.1 on the Evers et al. (2016) data sets and at 1.0 on the Sanson et al. (2018) data sets. log2 fold-change values calculated by CB2 were used as an input of RIGER.
RSA
We used the Python implementation of RSA (version 1.9, https://admin-ext.gnf.org/publications/RSA/). log2 fold-change values calculated by CB2 were used as an input of RSA.
sgRSEA
The sgRSEA R package (version 0.1, https://cran.r-project.org/web/packages/sgRSEA/) was used for benchmarking. We set the multiplier at 30.
PBNPA
The PBNPA R PACKAGE (version 0.0.2, https://cran.r-project.org/web/packages/PBNPA/) was used for benchmarking. We set the sim.no parameter at 10.
Specificity measure at sgRNA level
The specificity of detecting true-negative from the negative control sgRNAs is measured using 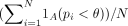 , where N is the number of nontargeting sgRNA, pi is the estimated p-value of the i-th sgRNA, θ is the p-value threshold, and 1A is the indicator function.
, where N is the number of nontargeting sgRNA, pi is the estimated p-value of the i-th sgRNA, θ is the p-value threshold, and 1A is the indicator function.
Algorithm for quantifying sgRNA abundance
Previous sgRNA abundance quantification methods
Recently published tools for CRISPR pooled screen analysis, including CRISPRcloud (Jeong et al. 2017), MAGeCK (Li et al. 2014), CRISPRAnalyzeR (Winter et al. 2017), and PinAPL-Py (Spahn et al. 2017), provide different methods for estimating the abundance of sgRNAs in each sample from pooled libraries. In most cases, input data consist of raw FASTQ-format sequencing result files.
CRISPRcloud was the first tool to offer an online user-defined, light-weight quantification method that proceeds on the user-client side. In contrast, CRISPRAnalyzeR and PinAPL-Py run their quantification methods on the server-side. As a result, CRISPRcloud minimized information passed through the Internet by transferring only the processed count matrix to the cloud storage.
However, as pointed out by Spahn et al. (2017), CRISPRcloud quantification algorithm can produce erroneous mapping results if the sgRNA sequences are staggered, because CRISPRcloud extracts sgRNA sequence for each read at a fixed location. Another limitation of CRISPRcloud is the fact that the user must decide where the extraction site is. Nevertheless, CRISPRcloud did not require tuning and was thus arguably more user-friendly than other tools. For instance, in PinAPL-Py, users need to set many tuning parameters for sgRNA quantification: adapter error rate parameter for trimming, matching and ambiguity thresholds, and parameters for alignment seed for the Bowtie alignment (Spahn et al. 2017).
We engineered CB2 to address precisely these issues. As a result, users no longer need to perform complicated parameter tuning for the sgRNA abundance quantification; one must simply provide the input files to CB2.
The binary representation of sgRNA sequence lowers the cost of computation
We used a binary representation for sgRNA sequence. This approach is memory efficient and improves the user experience at the client side (Melsted and Pritchard 2011). It only needs max(K, 2M) bits to store an sgRNA-sequence, where M is the length of the sequence, and K is the bit size to store a primitive integer in the machine (usually 64 bits) because we only need two bits to save a nucleotide (i.e., “A” is “00,” “C” is “01,” “G” is “11,” and “T” is “10”). The memory size is about half of that required for storing a character string of the sequence; that is, 160 bits are needed to store a 20-nt sgRNA sequence. Another benefit of binary representation is that it lowers the time complexity for the shift operator when comparing all k-mers of an sgRNA read using a sliding window. This is an essential function for the quantification algorithm in CB2. Compared with the string shift operator functions, such as string copy, substring extraction, and concatenation, the binary representation produces significantly shorter running times. Algorithm 2 in Supplemental Methods illustrates how a sgRNA library converted to a bit sequence and stored the converted sequence into a hash table.
Sliding window–based algorithm gives a high-resolution quantification with comparable running time
With a binary representation, we run the quantification algorithm as follows: First, we build a hash table for the reference library, with each key of the library in the hash table converted to the binary representation. Second, for each read, we scan the sequence of the read from 5′ to 3′ with the sliding window. In the ith iteration, the sliding window contains a substring of the read sequence from i to i + k − 1, where k is the length of the sgRNAs. The substring is also converted to a binary sequence, and the hash table is quickly checked to see if the sequence in the sliding window exists in the reference library. If the sequence is found in the hash table, then the count of the sequence is increased by one and the algorithm proceeds to the next read. Otherwise, it moves to i + 1-th iteration and the bit-shift method will be applied to take the next sliding window. Algorithm 1 in the Supplemental Methods represents a procedure of the sliding window–based algorithm.
For the case of a reverse complement sequenced sample, the entire procedure is repeated on the reverse complement reference sgRNA library, and the reads are scanned from 3′ to 5′. After both assays are performed (5′ to 3′ and 3′ to 5′ with the reverse complement reference sequences), mapping results between both sequences are compared. The one with a larger count corresponds to the correct sequence mapping. We compared the mappability of CB2 to those of MAGeCK (Li et al. 2015) and PinAPL-Py (Spahn et al. 2017) across multiple data sets from previous studies (Fig. 5).
Software availability
CRISPRBetaBinomial or CB2 is available as Supplemental Code and at https://CRAN.R-project.org/package=CB2. All of the data and scripts for the benchmarking are available in the Supplemental Material and at GitHub (https://github.com/hyunhwaj/CB2-Experiments). Parameters used in these experiments are described above in the Methods. CRISPRcloud is available at http://crispr.nrihub.org.
Acknowledgments
This work has been supported by National Institute of General Medical Sciences R01-GM120033, National Science Foundation–Division of Mathematical Sciences DMS-1263932, Cancer Prevention and Research Institute of Texas RP170387, Houston Endowment, the Hamill Foundation, and Chao Family Foundation (Z.L.), Huffington Foundation, Howard Hughes Medical Institute (H.Y.Z.), and the Parkinson's Foundation Stanley Fahn Junior Faculty Award PF-JFA-1762 (M.W.C.R.). We thank V.L. Brandt for editing the manuscript.
Author contributions: H-H.J., M.W.C.R., H.Y.Z., and Z.L. designed the study. H-H.J. and S.Y.K. implemented the CB2 software. H-H.J. and Z.L. performed analysis. Z.L. supervised the project. H-H.J., M.W.C.R., and Z.L. wrote the manuscript with input from all the authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245571.118.
-
Freely available online through the Genome Research Open Access option.
- Received October 23, 2018.
- Accepted April 3, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








