
The peopling of South America and the trans-Andean gene flow of the first settlers
- Alberto Gómez-Carballa1,2,3,8,
- Jacobo Pardo-Seco1,2,3,
- Stefania Brandini4,
- Alessandro Achilli4,
- Ugo A. Perego4,
- Michael D. Coble5,
- Toni M. Diegoli6,7,
- Vanesa Álvarez-Iglesias1,2,
- Federico Martinón-Torres3,
- Anna Olivieri4,
- Antonio Torroni4 and
- Antonio Salas1,2,8
- 1Unidade de Xenética, Departamento de Anatomía Patolóxica e Ciencias Forenses, Instituto de Ciencias Forenses, Facultade de Medicina, Universidade de Santiago de Compostela, 15782 Galicia, Spain;
- 2GenPoB Research Group, Instituto de Investigaciones Sanitarias (IDIS), Hospital Clínico Universitario de Santiago, Santiago de Compostela, 15706 Galicia, Spain;
- 3Grupo de Investigación en Genética, Vacunas, Infecciones y Pediatría (GENVIP), Hospital Clínico Universitario and Universidade de Santiago de Compostela, 15706 Galicia, Spain;
- 4Dipartimento di Biologia e Biotecnologie, Università di Pavia, 27110 Pavia, Italy;
- 5Applied Genetics Group, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA;
- 6Office of the Chief Scientist, Defense Forensic Science Center, Ft. Gillem, Georgia 30297, USA;
- 7Analytical Services, Incorporated, Arlington, Virginia 22201, USA
-
↵8 These authors contributed equally to this work.
Abstract
Genetic and archaeological data indicate that the initial Paleoindian settlers of South America followed two entry routes separated by the Andes and the Amazon rainforest. The interactions between these paths and their impact on the peopling of South America remain unclear. Analysis of genetic variation in the Peruvian Andes and regions located south of the Amazon River might provide clues on this issue. We analyzed mitochondrial DNA variation at different Andean locations and >360,000 autosomal SNPs from 28 Native American ethnic groups to evaluate different trans-Andean demographic scenarios. Our data reveal that the Peruvian Altiplano was an important enclave for early Paleoindian expansions and point to a genetic continuity in the Andes until recent times, which was only marginally affected by gene flow from the Amazonian lowlands. Genomic variation shows a good fit with the archaeological evidence, indicating that the genetic interactions between the descendants of the settlers that followed the Pacific and Atlantic routes were extremely limited.
The study of the human settlement and spread into the American double continent has received great attention in the literature (Torroni et al. 1993; Forster et al. 1996; Schurr and Sherry 2004; Tamm et al. 2007; Achilli et al. 2008; Fagundes et al. 2008; Gilbert et al. 2008; Perego et al. 2009, 2010; O'Rourke and Raff 2010; Bodner et al. 2012; Reich et al. 2012; Achilli et al. 2013; Raghavan et al. 2015; Llamas et al. 2016). It is generally agreed that the ancestors of Native Americans were probably of southern Siberian origin. The remains found at the Yana site represent unequivocal evidence of the Homo sapiens presence in the Arctic region ∼30 thousand years ago (kya), shortly before the Last Glacial Maximum (LGM) (Pitulko et al. 2004). However, recent archaeological evidence revealed human presence in the Arctic much earlier than previously thought (∼45 kya; SK mammoth and Bunge-Toll sites) (Pitulko et al. 2016). During the LGM, humans survived in glacial refuge areas (Torroni et al. 2001; Achilli et al. 2004; Gamble et al. 2004; Gómez-Carballa et al. 2012; Pala et al. 2012). One of these was Beringia, a land bridge located between the northeastern continental region of Asia and the northwestern extreme of North America, which was largely unglaciated during the Pleistocene. During the LGM, the ancestors of Paleoindians remained almost isolated, possibly up to 6–8 ky, in eastern Beringia (Raghavan et al. 2014, 2015; Llamas et al. 2016) or, as paleoecological data much more strongly indicate, in south-central Beringia (Elias and Crocker 2008; Hoffecker et al. 2014, 2016). In that period of global cooling and glacial advance, genetic drift, novel mutations, and limited admixture with newly arrived East Asian groups extensively reshaped their genetic variation (Perego et al. 2009, 2010).
Genetic and archaeological data support the hypothesis that a small population group entered the American continent from Beringia 15–18 kya, in concomitance with the improvement of climatic conditions (Achilli et al. 2008; Goebel et al. 2008; Perego et al. 2009; Bodner et al. 2012; Battaglia et al. 2013; Llamas et al. 2016). More complex scenarios based on genetic and linguistic evidence envision instead two, three, or maybe more migration waves from Beringia (Torroni et al. 1992; Perego et al. 2009; Reich et al. 2012; Achilli et al. 2013). The initial incursion of Paleoindians into America was followed by a southward expansion wave along the ice-free Pacific coastal line (Tamm et al. 2007; Fagundes et al. 2008; Bodner et al. 2012), which crossed Mesoamerica and reached the northern latitudes of South America. Here, Paleoindians split—one subset followed the Pacific and colonized the Andean region, and another one expanded along the Atlantic (Reich et al. 2012). The rapid coastal migration movements played a major role in the colonization of the whole South American subcontinent, and genetic age estimates fit well with the archaeological evidence of human presence in the Southern Cone at the Monte Verde site (Chile) at least ∼14.5 kya (Dillehay 1989; Dillehay et al. 2015; Brandini et al. 2018).
The initial isolation period in Beringia caused both the loss and emergence of maternal lineages, affecting local haplogroup frequencies, as well as the molecular differentiation from their East Asian ancestors. At least 16 Asian or Beringian mtDNA founders contributed to the initial settlement of America (Achilli et al. 2008, 2013; Perego et al. 2010; Hooshiar Kashani et al. 2012). As recorded in both autosomal (Reich et al. 2012; Llamas et al. 2016) and uniparental (Torroni et al. 1993; Bodner et al. 2012; de Saint Pierre et al. 2012b; Roewer et al. 2013) markers, shortly after the initial entry into the double continent, populations began to diverge, favored by small effective population sizes and long isolation periods, generally interrupted by only brief periods of limited gene flow. Phylogeographic analyses of mtDNA variation have shed light on large-scale demographic aspects of the Paleoindian colonization but also on minor events that took place in the continent during the initial spread (Perego et al. 2009, 2010; Bodner et al. 2012; de Saint Pierre et al. 2012a,b; Achilli et al. 2013). The analysis of geographically restricted sub-haplogroups added further support to the scenario of an initial split of the first Paleoindian settlers of South America into two main groups whose expansion routes followed different paths—along the Pacific and the Atlantic coastlines—and highlighted the possibility of trans-Andean events of gene flow at different latitudes (Bodner et al. 2012). Finally, there is recent mtDNA evidence that present-day Peru and Ecuador were first populated by Paleoindian settlers of North American ancestry in the short time frame of 16.0–14.6 kya (Brandini et al. 2018). Despite this recent insight, the colonization process and subsequent population movements that led to the peopling of South America remain poorly known.
The aim of the present study is to shed light on the demographic movements that occurred in the region after the initial colonization wave, to identify and characterize trans-Andean gene flow, and to explore its implications at the continental level.
Results
Phylogeography of Andean mtDNA lineages
Haplogroup B2 represents ∼23% of the mtDNAs in South America, but its frequency varies across the subcontinent (Salas et al. 2009). In the Andean region, B2 ranges from moderate frequencies in the general population from Ecuador (20%) and in some Ecuadorian Natives, e.g., Cayapa (40%) (Rickards et al. 1999), to very high frequencies (up to 90%) in some Quechua and Aymara populations in the Andean regions of Bolivia and Peru (Bert et al. 2001; Barbieri et al. 2011; Gayà-Vidal et al. 2011). Several selected B2 sub-haplogroups were investigated here in order to further characterize their internal variation and shed light on the Paleoindian settlement of the South American continent.
We named as B2ai a new B2 sub-haplogroup that is defined by the stable transition C16168T (phylogenetic weight = 5.6 in PhyloTree 17 [Weissensteiner et al. 2016]) on top of the B2 mutational motif (Fig. 1A). B2ai appears almost exclusively in the Peruvian and Bolivian Andes, with approximately the same frequency in both Peru and Bolivia (∼4%) (Fig. 1B), and it probably arose in this area ∼8.4 kya (7.8–9.1 kya) (Fig. 1C), where the molecular diversity indices are also high (Supplemental Table S1). Control-region (CR) networks show a star-like pattern in both regions, mirroring recent population growth. There are two ancient mitogenomes from Peru within B2ai, one of them belonging to B2ai2a subclade (that contains one Argentinean and one Bolivian mitogenomes). CR sequences also provide further information (Fig. 1D): Haplogroup B2ai has spread into Ecuador and northern Chile. In ancient DNA samples, there are a total of two mitogenomes and 20 CR sequences (with moderate diversity), all of them from Peru.
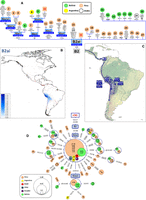
(A) Maximum parsimony tree of B2ai complete mitogenomes. ML coalescence ages (in ky) are shown below haplogroup names. (B) Interpolated frequencies of B2ai mtDNAs in America; red dots indicate sampling points. (C) Main migration routes of B2ai as inferred from phylogeographic data. (D) Network of B2ai CR haplotypes obtained from population surveys (listed in Supplemental Table S2). The position of the revised Cambridge reference sequence (rCRS) is indicated for reading sequence motifs. In panels A and D, mtDNA mutations are indicated along the branches of the phylogenies; they are transitions unless a suffix A, C, G, or T is included to indicate a transversion. Other suffixes indicate: insertions (+), synonymous substitutions (s), mutational changes in tRNA (-t), mutational changes in rRNA (-r), and noncoding variants located in the mtDNA coding region (-nc). Amino acid replacements are in round brackets, underlined mutations indicate homoplasies in the same tree, while the prefix “@” indicates a back mutation. Haplotypes from ancient samples are in boxes (and red numbers), while haplotypes from present-day samples are in circles. Geographic and/or ethnic origins of the mitogenomes in panel A are provided in Supplemental Table S5, while the two-letter codes inside circles in panel D refer to ethnic origins (according to Supplemental Table S2), with numbers indicating the number of mtDNAs. Boxes with blue borders in panel A indicate sub-haplogroups that are new relative to the reference phylogeny in PhyloTree Build 17. Mutational hotspot variants at positions 16182, 16183, and 16519, as well as variation around position 310 and length or point heteroplasmies were not considered for the phylogenetic reconstruction. Mitochondrial DNA sequence data for comparisons, phylogenetic trees, and maps of interpolated frequencies were obtained from the literature (Supplemental Table S10).
We identified a second new B2 subclade, B2aj (motif A8853G–G15883A–C16188T) (Fig. 2). Its molecular divergence suggests an origin slightly earlier than B2ai, ∼10.4 kya (9.1–11.8 kya). According to mitogenomes and CR sequences, B2aj is mainly distributed in Peru and Bolivia, but it is also abundant in the Argentinean Andes. A Peruvian origin for B2aj can be postulated, taking into account that its molecular diversity is highest in Peru (HD = 0.7530; π = 0.0047) relative to other Andean countries, where, while this lineage is found at a higher frequency (e.g., 20.4% in Bolivia vs. 10.8% in Peru), it shows much lower diversity (e.g., Bolivia HD = 0.5227; π = 0.0026) (Supplemental Table S1). The network of B2aj CR sequences (Fig. 2B) is clearly star-like, suggesting an important Ne growth in the Andes, especially in Bolivia (except around the Titicaca basin, which is close to the present-day border with Peru; see below). B2aj spread most successfully into north Andean Argentina probably because of its high frequency in Bolivia. The interpolated frequency map shows that B2aj is almost exclusively present in the Andean region plus the above-mentioned Argentinean component (Fig. 2C). One of its subclades (B2aj1a1a; time to the most recent common ancestor [TMRCA]: 0.8 kya) might have originated recently in Argentina (Fig. 2A). Overall, phylogenetic patterns suggest that the gene flow of B2aj was more favored southeastward (Bolivia to Argentina) than in the opposite direction (Bolivia to Peru) (Fig. 2D). Argentinean mitogenomes share several B2aj subclades with Bolivian samples, and these common subclades have TMRCAs ranging from 1.7 (B2aj4a2) to 5.2 (B2aj3c) kya, suggesting a continuous gene flow between these neighboring Andean regions. Ancient B2aj samples were found in the Titicaca region (Tiwanaku; dated 0.8 kya) and in the northwestern Argentinean province of Jujuy (Quebrada and Puno; dated ∼0.6–1 kya).
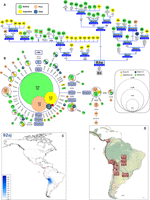
(A) Maximum parsimony tree of B2aj complete mitogenomes. (B) Network of B2ab CR haplotypes obtained from population surveys (listed in Supplemental Table S2). (C) Interpolated frequencies of B2aj mtDNAs in America. (D) Main migration routes of B2aj as inferred from phylogeographic data. See the legend of Figure 1 for more details on the phylogenies of panels A and B.
B2y mtDNAs are defined by the transition C16261T (phylogenetic weight = 4.2 in PhyloTree 17 [Weissensteiner et al. 2016]) (Fig. 3). This is an ancient clade that most likely arose along the Pacific coast of North America ∼14.3 kya (13.0–15.7 kya) during the initial settlement of the continent. This scenario is supported by the extent of diversity from the root CR haplotype observed in Native samples from the US (Fig. 3C; Malhi et al. 2003; Kemp et al. 2010). The phylogeographic pattern of B2y suggests that its founding haplotype most likely entered South America following an expansion along the Pacific coast that reached northern Chilean latitudes. B2y is only marginally found in French Guiana (Mazieres et al. 2011) and in the Gavião (Brazilian Amazon forest [Ward et al. 1996]) (Fig. 3B). The interpolated frequency map of B2y lineages (Fig. 3D) shows a frequency peak in North America, high frequencies in the Andean area, and a moderate presence in north Argentina. The subclade B2y2a1a most likely arose in Peru 6.6 kya (5.3–7.9 kya) (Fig. 3A,C) and later spread into other Andean locations (Bolivia), and in particular into the Gran Chaco (northeastern Argentina) (Cabana et al. 2006; Sevini et al. 2013), which was reached by passing through the Jujuy province (northwestern Argentina) (Cardoso et al. 2013). Ancient B2y DNAs were exclusively found in Peruvian samples.
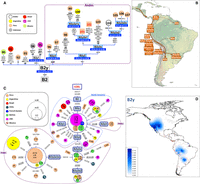
(A) Maximum parsimony tree of B2y complete mitogenomes. The B2y Andean sublineages (B2y2 and B2y3) are highlighted in the dotted rectangle. (B) Main migration routes of B2y as inferred from phylogeographic data. (C) Network of B2y CR haplotypes obtained from population surveys (listed in Supplemental Table S2). (D) Interpolated frequencies of B2y mtDNAs in America. See the legend of Figure 1 for more details on the phylogenies of panels A and C.
B2b is defined by the transition G6755A and probably arose ∼16.6 kya (15.6–17.5 kya), a little earlier than B2y. Phylogeographic patterns of B2b mtDNAs suggest that this clade most likely arose in the north of Mesoamerica (Brandini et al. 2018). The greatest proportion of mitogenomes are from South America (93.3%), mostly from the Andean region, especially from Peru (46.6%) (Fig. 4A,B). It seems that B2b successfully spread through both the Pacific and the Atlantic coasts. The Pacific route was probably led by B2b11 ∼14.7 kya (13.5–15.9 kya), with B2b14 expanding slightly later (12.8 kya [11.0–11.6 kya]); both most likely initially settled in Peru as indicated by the high CR diversity values relative to surrounding regions (e.g., Bolivia) (Supplemental Table S1). CR data show that several B2b14 haplotypes are shared between Peruvians and Bolivians, suggesting intense gene flow at these latitudes of the Andes; a few B2b14 mtDNAs were also detected in northern Argentineans, with a singleton in Buenos Aires that might signal recent migration (Fig. 4B,C,E). In addition, four CR B2b14 sequences from ancient specimens were found in Pacapaccari, a location in the fringe area between the Andean highlands and the Pacific coast of Peru.
(A) Maximum parsimony tree of Bb3, B2b11 and B2b14 complete mitogenomes. An asterisk as prefix indicates a position located in an overlapping region shared by two mtDNA genes. For the sake of clarity, the mitogenomes belonging to B2b (× B2b3, B2b11, B2b14) are simply indicated as rectangles in the left side of the tree without accounting for their phylogeny (listed in Supplemental Table S11); the colors of rectangles indicate the geographic origin (as shown in the legend), and the inner numbers correspond to the number of detected mitogenomes. (B) Phylogenetic network of B2b3, B2b11, and B2b14 CR haplotypes obtained from population surveys (listed in Supplemental Table S2). (C) Main migration routes of B2b, B2b3, B2b11, and B2b14 as inferred from phylogeographic data. (D) Interpolated frequencies of B2b3 mtDNAs in America. (E) Interpolated frequencies of B2b14 mtDNAs in America. (F) Interpolated frequencies of B2b11 mtDNAs in America. See the legend of Figure 1 for more details on the phylogenies of panels A and B.
The geographic features of B2b11 are slightly different from those of B2b14. B2b11 is mainly restricted to the central regions of Peru, with indications of limited gene flow southward, reaching the highlands of Cusco and Andahuaylas in southern Peru (Fig. 4F). Ancient B2b11 samples are only found along the coast of central Peru (Huaca Pucllana and Pueblo Viejo) (Fig. 4A). It is likely that B2b11 originated in the central coastal regions of Peru in close association with the Wari, Ychsma, and Inca cultures that sequentially occupied that area from AD 600 to 1500 (Supplemental Table S2).
The age of the B2b3 branch (15.8 ky) indicates that it probably played a part in the pioneering migration wave that moved southward from Mesoamerica and then along the Atlantic facade of South America. The colonization of the Atlantic coast by B2b3a carriers occurred later than their B2b Andean counterparts, ∼8.3 kya (7.0–9.5 kya). The phylogeny based on the CR data shows clear signals of population expansion in the region with at least one main founder haplotype (Fig. 4B). The origin of B2b3a (and B2b3a1, too) could be placed close to the Amazon River delta, as witnessed by its high frequency in the region (Fig. 4D) as well as the detection of a 4-ky-old sample from Pirabas (north of Pará state) carrying the HVS-I motif T16189C–T16217C–T16249C–A16312G–C16344T (Ribeiro-dos-Santos et al. 1996). The subclade B2b3a2 arose in Brazil ∼3.9 kya (3.3–4.8 kya) and likely spread southward following the Atlantic as testified by the presence of a B2b3a2 member in Uruguay (Guarani) (Sans et al. 2015) and Argentineans from Río de la Plata (Fig. 4B). The subclade B2b3a1 appeared earlier (6.9 kya [5.5–8.2]), probably in Brazil, although there is only one mitogenome in Uruguay representing its phylogenetic root (Fig. 4A).
Most interesting is the derived sublineage B2b3a1a that clearly moved northward from Venezuela/Brazil to the Caribbean ∼6.0 kya (5.1–6.8 kya) and solidly settled in Puerto Rico (Fig. 4B), representing 100% of the B2 haplotypes found on this island (The 1000 Genomes Project Consortium 2012; Vilar et al. 2014). Outside Puerto Rico, there are only members of B2b3a1a on the northern coast of Venezuela (probably a crossing point to the Caribbean from Brazil) (Castro de Guerra et al. 2012; Brandini et al. 2018), in Cuba (Mendizabal et al. 2008), and the Dominican Republic (Tajima et al. 2004). The two mitogenomes and one CR sequence sampled in the US (Fig. 4A,B) belonging to B2b3a1a might represent a recent arrival, probably from Puerto Rico, as recently suggested (Brandini et al. 2018).
Bayesian-based inferences of gene flow in the Andes
Of the nine demographic models tested, the one that received the highest probability (0.825) was the Stepping Stone linear model: Peru → Bolivia → NW-Argentina, followed by a linear model (probability = 0.175) that additionally considered bidirectional gene flow between Bolivia and Argentina (Peru → Bolivia ↔ NW-Argentina). However, according to the latter model, the number of migrants involved in a south to north movement from northwest Argentina to Bolivia would have been very low (Supplemental Table S3).
Gene flow models involving populations from Andean Bolivia and northern Argentina (NW-Argentina and NE-Argentina) (Supplemental Table S4) were also evaluated. Again, the unidirectional gene flow Bolivia → NW-Argentina → NE-Argentina was the best fitting model (probability = 1.0). We also observed twice as high gene flow for Bolivia → NW-Argentina than for NW-Argentina → NE-Argentina.
Genome-wide autosomal data and trans-Andean gene flow
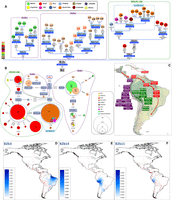
More than 360K autosomal SNPs were analyzed in Native South Americans in order to shed light into the role played by the Andes in the demography of the subcontinent. We first computed identity-by-state (IBS) and FST distances between Peruvians (PEL-1000G) and the rest of southern Native Americans. An initial multidimensional scaling (MDS) plot of IBS values showed South Americans clustering together when plotted against populations from Europe and Africa (Supplemental Fig. S1). When exploring only South American samples in a MDS plot, two populations appeared as genetic outliers, the Surui and the Karitiana (Supplemental Fig. S2); for this reason, they were removed from subsequent analyses. Dimension 1 of the MDS in Figure 5A clusters all Natives from the Andes (Quechua, Peru, Aymara, and Diaguita) on one side of the plot, supporting their close relationships but also their differentiation or demographic isolation from the other native groups. Dimension 2 shows at one pole of the plot the Southern Cone populations (Yaghan, Chilote, Chono, and Hulliche) and at another pole those living in the northern extreme of the subcontinent (Arhuaco and Kogi), whereas the remaining populations scatter in between.
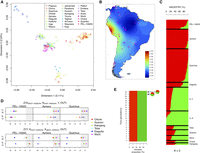
(A) MDS based on IBS values obtained from different Native American populations from South America. (B) Interpolated FST values between Peruvian and other Native American samples. (C) Admixture analysis for the best cross validation value K = 2, showing the admixture components shared between different populations from South America. (E-T) Equatorial-Tucanoan, (C-P) Chibchan-Paezan. (D) D-statistics computed to formally test for admixture between West-Andean and East-Andean populations. (E) Time of the main admixture event occurring in the Diaguita between an ancestral component from Equatorial-Tucanoan (green) populations located to the east of the Andes and Peruvian variation (red).
Figure 5B shows a map of interpolated FST values between Peruvians and other Native American samples from all across South America, highlighting the close genetic proximity between Peruvians and neighboring Andean populations as well as the relevant genetic distances with the rest of the subcontinent. The map of interpolated IBS values shows the same pattern (Supplemental Fig. S3).
Admixture analysis consistently suggests a shared ancestry between all Andean populations (Fig. 5C). IBS distances between Peru and other populations in South America were also geographically interpolated in order to visualize the relationships between Andean populations and the rest. The map (Fig. 5B) displays a virtual genetic continuity in the Andes indicating a substantial genetic isolation of this mountainous area from the rest of the South American native populations.
Both mtDNA and autosomal data point to a limited trans-Andean genetic diffusion from Peru (and neighboring populations) toward the east of South America. In order to formally test this hypothesis, we first defined population sets as follows:
-
XWEST-ANDEAN: represents populations PEL-1000G, Aymara, and Quechua, who live on the western side of the Andes;
-
Y: represents the Chibchan-Paezan and Equatorial-Tucanoan linguistic families of the eastern path of South American spread;
-
XEAST-ANDEAN: represents the populations that are located in between XWEST-ANDEAN and Y-populations; that is, populations such as the Chane, Guarani, Kaingang, Toba, Diaguita, and Wichi, who live immediately to the east of the Andes mountain range at subAmazon latitudes (it is at this latitude that genetic exchange could have been most favored).
Then, we computed D-statistics as follows:
-
D(Y, XEAST-ANDEAN, XWEST-ANDEAN, OUT): statistically tests if there was gene flow between XEAST-ANDEAN populations and populations Y (East South America) (Fig. 5D).
-
D(XWEST-ANDEAN, XEAST-ANDEAN, Y, OUT): tests if there was gene flow between XEAST-ANDEAN and XWEST-ANDEAN (Fig. 5D).
The analyses indicate that the Diaguita (NW-Argentina) is the only population of northern Argentina with a statistically significant input from populations living in the Peruvian/Bolivian Andes (Aymara and Quechua).
Furthermore, all populations living on the eastern side of the Andes (northern Argentina: Wichi, Diaguita, Toba, Chane, etc.) show a statistically significant contribution from populations representing the eastern path of the South American spread when using PEL-1000G as a reference population, a signal that is diluted when using Aymara and Quechua.
Dating the time of admixture between Andean Natives and Diaguita
Although diffusion of genetic variation from the Andes into southern latitudes has been limited, statistical models indicate that there was moderate gene flow with populations living in NW-Argentina, the Diaguita. The track length distribution of genomic segments inherited from different ancestral populations allows dating of the admixture time. This analysis suggests that a main admixture event involved the ancestors of modern Diaguita, with an Equatorial-Tucanoan component from the southern latitudes of South America (using Guahibo, Wayuu, Piapoco, Guahibo, Palikur, and Parakana as surrogate population sets) and a Peruvian (PEL-1000G) component, an event that occurred in two main pulses ∼21 and 22 generations ago (525–550 ya; using 25 yr per generation) (Fig. 5E).
Discussion
Phylogeographic data inferred from mitogenomes highlight the demographic importance of the Andean region during the South American settlement process and suggest a history of evolutionary success and high-altitude adaptation. Goldberg et al. (2016) have recently reconstructed the spatiotemporal patterns of human population growth in South America using archaeological information and suggested two main demographic periods: (1) an initial one involving a rapid colonization but low Ne and lasting ∼8 ky; and (2) a period of population growth along the Pacific coastline (in Peru, Chile, and Ecuador) beginning ∼5 kya and coinciding with the Neolithic transition (and the onset of sedentism). In Figure 6, we show the location of the mDNA lineages analyzed in the present study together with the interpolated archaeological data from Goldberg et al. (2016) (see also Fig. 1 of Goldberg et al. 2016). The map shows the great fit between archaeological and mtDNA evidence, then suggesting that B2 is a good marker of the demographic transformation occurring in the Andes. The highest frequencies of B2 in South America are by far in the Andean regions of Peru, Bolivia, and northern Argentina, reaching frequencies up to 70%–90% in some Aymara and Quechua Andean populations (Bert et al. 2001; Barbieri et al. 2011; Heinz et al. 2013, 2015). The pre-Columbian mtDNA data available follow the same pattern: B2 reaches high frequencies in the Peruvian highlands, ranging from 42% to 75% (Shinoda et al. 2006; Baca et al. 2012, 2014). Ancient and contemporary data indicate a genetic continuity in the Central Andes that is compatible with a high Ne and intense gene flow in this area for a long time period (8–9 ky), with limited introgression from surrounding regions (Batai and Williams 2014; Cabana et al. 2014; Fehren-Schmitz et al. 2017). Moreover, the observed genetic patterns in the Central Andes can be tentatively linked to the population expansions of the most relevant Andean empires: the Tiwanaku, the Wari, and more recently, the Incas (Fehren-Schmitz et al. 2014).
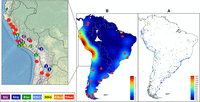
(A) Age in years (see colored legend inset) and location of archaeological sites in South America according to Goldberg et al. (2016). (B) Interpolated kernel density map (units are expressed as the square decimal degree) from the archaeological data in Goldberg et al. (2016); dots indicate the sampling points of ancient specimens and the availability of mtDNA data. While red dots indicate mtDNA lineages other than those analyzed in the present study, other colors signal the presence and number of the B2 lineages analyzed here (see the legend inset).
The good fit between archaeological and genetic data finds further support in the EBSPs of Ne population growth. B2ai shows a sudden increase of Ne starting ∼10 kya, while South American B2y and B2aj show a more progressive growth in a recent time range (from 4 to 6 kya). These lineages are located almost exclusively in the Andes and probably emerged and diversified in the Andean area soon after the initial colonization wave (Fig. 7A). While population growth inferred from B2ai suggests the existence of an important population expansion starting already during the Andean Preceramic periods II to III (a fact that is not highlighted in the archaeological record) (Fig. 7A), lineages such as South American B2y and B2aj experienced a great population growth starting ∼5 kya, thus perfectly matching the beginning of the major demographic expansion in the Andes.
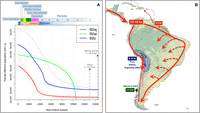
(A) EBSP analysis of the mtDNA lineages explored in the present study. The figure shows the population growth experienced during the main prehistorical periods of the South American settlement. The EBSP of B2y considers only mtDNAs observed in South America. (B) Demographic scenario for the peopling of South America as suggested by the results of the present study.
Autosomal data satisfy this demographic scenario, indicating a strong genetic affinity between the populations of the Andean region and their isolation with respect to other American regions (Fig. 5).
Furthermore, our data also clarify the admixture patterns occurring immediately to the east of the Andes at northern Argentinean latitudes. The genome of present-day Diaguitas seems to be the consequence of a single event or multiple admixture events that occurred ∼525–550 ya between a Peruvian-Central Andean component and an ancestral one of Equatorial-Tucanoan origin. The time estimate agrees remarkably well with the known expansion of the Incas into Diaguita territories occurring >500 ya, at the time of the Inca emperor Túpac Inca Yupanqui (Diaguita is most likely a Quechua voice imposed by the Incas) (Dillehay and Netherly 1998).
By integrating all the available evidence, it is possible to conceive a demographic model for the peopling of South America (Fig. 7B). A few waves of Paleoindians could have reached the northern edge of South America ∼16–15 kya. These settlers advanced across two main colonization routes, following the Atlantic and the Pacific coasts. The populations that settled the Peruvian Andes were demographically very successful and experienced rapid population growth from at least 8 kya to the present day, which led to the incubation of important genetic variation in the highlands. Substantial gene flow existed in the territory spanning present-day Peru and Andean Bolivia. Population movements were most favored in a north to south direction, not bidirectionally. The Paleoindians living at this latitude of the Andes had very limited genetic exchange with the East of the subcontinent and even with populations living to the south (with the exception of those that settled the narrow Andean fringe running along the present-day Chilean territories). Probably, the Amazon forest acted as a geographical barrier to gene flow between the Pacific and the Atlantic expansion waves. The Atlantic migration wave played a major role in the colonization of the non-Andean regions. Although demographic growth was likely more moderate here than in the Peruvian Andes in ancient times, it might have been more relevant in more recent times, as suggested by the Ne estimated from the Brazilian B2b3 clade, with a quick growth starting 2.5 kya (Supplemental Fig. S4). At this time, population movements along the Atlantic coast could have been important not only in a north to south direction but also with an opposite component that might have even involved movements toward the Caribbean. It is haplogroup B2b3a1a that best testifies to this continental-Caribbean connection. Thus, while the ancestral nodes of B2b3 were found on the Atlantic coast of South America (reaching at least Uruguay), we observed the derived B2b3a1a sub-haplogroup in Puerto Rico at relatively high frequencies (here, 100% of B2 haplotypes are B2b3a1a, representing 8.3% of the Native American component), most likely arriving there ∼6 kya (almost ∼10 kya after the occurrence of B2b3 in continental South America). B2b3a1a could have been part of the Taino's legacy in the modern Caribbean population, representing an ancestral northern South American input in the Greater Antilles. In turn, this lineage possibly arose (from an ancestral Brazilian node) around the Orinoco Basin (Venezuela) not earlier than 6 kya. Alternatively, B2b3a1a could have arisen locally in the Tainos from Puerto Rico, from where it could have spread across the Caribbean and to northern Venezuela. A recent paper on an ancient B2 Lucayan Taino sample from the Bahamas (calibrated to AD 776–992) raises the possibility of a pre-Columbian arrival of haplogroup B2 to the Greater Antilles (Schroeder et al. 2018). This Taino specimen shares ancestry with modern Puerto Ricans and is related to Arawakan speakers from northern South America.
Overall, genetic and archaeological data point to the Andes as the main demographic source for South America in prehistoric and historical times. The homogeneous ecological environment of the highlands most likely favored the incubation of prosperous civilizations and cultures, a complex set of circumstances that was not as favorable in other continental regions. Adaptation to high altitude might have pushed these populations to persist and prosper there. Such a scenario would explain the very limited interplay between the two main routes of colonization of the subcontinent and the scarce genetic exchange with other South American populations.
Methods
Samples, DNA extraction, and amplification
We selected a total of 146 mitogenomes from ancient and modern populations belonging to haplogroup B2 (Supplemental Table S5); 72 were newly sequenced in this study, while the rest were retrieved from GenBank (n = 59) (https://www.ncbi.nlm.nih.gov/genbank/) and from The 1000 Genomes Project (n = 15) (www.1000Genomes.org; hereafter, 1000G) using the GATK Toolkit from the 1000G repository and processed as in Gómez-Carballa et al. (2015). We also collected samples from natives living in the northernmost Andean provinces of Argentina: 45 Kollas from Salta and Jujuy, and 24 Diaguitas from Catamarca (Supplemental Table S6). DNA was extracted following standard phenol-chloroform protocols. Written informed consent was obtained under protocols approved by the Asociación de Bioquímicos de Córdoba (Argentina) and the Universidad Católica de Córdoba (Argentina).
In addition, we compiled 822 CR sequences from the literature belonging to the targeted B2 subclades (Supplemental Table S2). Sub-haplogroup affiliations were inferred from the available sequence data (generally HVS-I and/or HVS-II).
PCR amplification, sequencing, and SNP analysis were carried out following the protocols described in Supplemental Text.
Statistical analyses on mtDNA data
Diversity indices were computed as indicated in Supplemental Text.
mtDNAs were classified into haplogroups and checked for data quality using HaploGrep 2 (Weissensteiner et al. 2016). CR data from Supplemental Table S2 were used to build the networks in Figures 1–4 (sequence range nps 16090–16362). Subsequently, the maximum likelihood (ML) approach implemented in PAMLX 1.3.1 (Xu and Yang 2013) was used to estimate the time to the most recent common ancestor for the main phylogenetic nodes, as in Brandini et al. (2018). The MP phylogenetic tree was built referring to the haplogroup nomenclature of PhyloTree (van Oven and Kayser 2009). The 72 novel B2 mitogenomes, together with 570 other Native American mitogenomes previously employed in Brandini et al. (2018) (Supplemental Table S7), were used to estimate sub-haplogroup ages (Supplemental Table S8; details in Supplemental Text).
Extended Bayesian skyline plots and trans-Andean gene flow analysis were carried out as explained in Supplemental Text. Maps of interpolated frequencies were built as detailed in Supplemental Text.
Demography inferred from genome autosomal data
SNP data were compiled and intersected from two main genome repositories: Native American data available from Reich et al. (2012) and data from 1000G. SNP genotypes were masked for the Native American component using PCAdmix (Brisbin et al. 2012), and BEAGLE 3.3.2 (Browning and Browning 2007) was used to prepare the necessary SNP unphased and imputed data for PCAdmix. The final data set consisted of 362,765 SNPs representing Native American variation in a total of 28 South American ethnic groups (Supplemental Table S9). We initially computed identity-by-state values from SNP data using PLINK (Purcell et al. 2007). Multidimensional scaling was carried out on a matrix of pair-wise identity-by-state values, using the function cmdscale from the library stats from R (http://www.R-project.org/) (R Core Team 2011). Admixture components of Native American populations were estimated using ADMIXTURE (Alexander et al. 2009). D-statistics were computed to formally test for genetic admixture using ADMIXTOOLS (Patterson et al. 2012). An artificial outgroup was built as in Pardo-Seco et al. (2016). Timing of admixture was obtained from the analysis of tract length of chromosomal segments of different ancestries using the Tracts (Gravel 2012). IBS and FST distances were also displayed in geographic interpolated maps (Supplemental Text).
Data access
Newly generated mitogenome sequences from this study have been submitted to the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MG637053–MG637124.
Acknowledgments
We thank Juan Carlos Jaime for help, support, and access to samples. This study received support from the Instituto de Salud Carlos III (Proyecto de Investigación en Salud, Acción Estratégica en Salud): project GePEM ISCIII/PI16/01478/Cofinanciado FEDER) (to A.S.); project ReSVinext ISCIII/PI16/01569/Cofinanciado FEDER (to F.M.-T.); Consellería de Sanidade, Xunta de Galicia (RHI07/2-intensificación actividad investigadora, PS09749 and 10PXIB918184PR), Instituto de Salud Carlos III (Intensificación de la actividad investigadora 2007–2012, PI16/01569) (to F.M.-T.); Fondo de Investigación Sanitaria (FIS; PI070069/PI1000540) del plan nacional de I + D + I and ‘fondos FEDER’ (to F.M.-T.); 2016-PG071 Consolidación e Estructuración REDES 2016GI-1344 G3VIP (Grupo Gallego de Genética Vacunas Infecciones y Pediatría, ED341D R2016/021) (to A.S. and F.M.-T.); the University of Pavia strategic theme “Towards a governance model for international migration: an interdisciplinary and diachronic perspective” (MIGRAT-IN-G) (to A.A., A.O., and A.T.); the Italian Ministry of Education, University and Research: Progetti Futuro in Ricerca 2012 (RBFR126B8I) (to A.A. and A.O.); the Progetti Ricerca Interesse Nazionale 2012 (to A.A. and A.T.); Dipartimenti di Eccellenza Program (2018–2022)—Dept. of Biology and Biotechnology “L. Spallanzani,” University of Pavia (to A.A., A.O., and A.T.); and the ERC Consolidator Grant: CoG-2014, No. 648535 (to A.A.).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234674.118.
- Received January 14, 2018.
- Accepted April 27, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








