
Precise and efficient nucleotide substitution near genomic nick via noncanonical homology-directed repair
- Kazuhiro Nakajima1,5,
- Yue Zhou1,5,
- Akiko Tomita1,6,
- Yoshihiro Hirade1,6,
- Channabasavaiah B. Gurumurthy2,3 and
- Shinichiro Nakada1,4
- 1Department of Bioregulation and Cellular Response, Graduate School of Medicine, Osaka University, Suita, Osaka 565-0871, Japan;
- 2Mouse Genome Engineering Core Facility, Vice Chancellor for Research Office, University of Nebraska Medical Center, Omaha, Nebraska 68198, USA;
- 3Developmental Neuroscience, Munroe-Meyer Institute for Genetics and Rehabilitation, University of Nebraska Medical Center, Omaha, Nebraska 68198, USA;
- 4Institute for Advanced Co-Creation Studies, Osaka University, Suita, Osaka 565-0871, Japan
Abstract
CRISPR/Cas9, which generates DNA double-strand breaks (DSBs) at target loci, is a powerful tool for editing genomes when codelivered with a donor DNA template. However, DSBs, which are the most deleterious type of DNA damage, often result in unintended nucleotide insertions/deletions (indels) via mutagenic nonhomologous end joining. We developed a strategy for precise gene editing that does not generate DSBs. We show that a combination of single nicks in the target gene and donor plasmid (SNGD) using Cas9D10A nickase promotes efficient nucleotide substitution by gene editing. Nicking the target gene alone did not facilitate efficient gene editing. However, an additional nick in the donor plasmid backbone markedly improved the gene-editing efficiency. SNGD-mediated gene editing led to a markedly lower indel frequency than that by the DSB-mediated approach. We also show that SNGD promotes gene editing at endogenous loci in human cells. Mechanistically, SNGD-mediated gene editing requires long-sequence homology between the target gene and repair template, but does not require CtIP, RAD51, or RAD52. Thus, it is considered that noncanonical homology-directed repair regulates the SNGD-mediated gene editing. In summary, SNGD promotes precise and efficient gene editing and may be a promising strategy for the development of a novel gene therapy approach.
Gene therapy, involving the correction of disease-causing genetic mutations, has been one of the most challenging tasks in medicine. Long DNA sequences homologous to a target locus and selectable marker genes have been used for gene targeting in cells. However, the efficiency of this gene-targeting approach is poor. Introducing a double-strand break (DSB) close to the target site can markedly increase the efficiency of gene targeting (Doudna and Charpentier 2014; Maeder and Gersbach 2016). Programmable nucleases, such as ZFNs, TALENs, and CRISPR/Cas9, have been used to generate DSBs for gene editing (Woolf et al. 2017). Among several systems available for generating a DSB at a target locus, CRISPR/Cas9 is the most widely used tool. The Cas9 protein forms a complex with CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA), or with a single-guide RNA (sgRNA), a fusion of crRNA and tracrRNA. This protein-RNA complex scans and detects the target sequence adjacent to the protospacer adjacent motif (PAM) in the genome. Once Cas9 is recruited to the target site, it cleaves each of the two strands of DNA through its HNH and RuvC-like nuclease domains. This generates a DSB at the site (Doudna and Charpentier 2014; Hsu et al. 2014).
DSBs are repaired by homology-directed repair (HDR) or nonhomologous end joining (NHEJ) (Goodarzi and Jeggo 2013). Homologous recombination (HR) is an error-free HDR, wherein a sister chromatid or exogenous DNA serves as a donor. During HR, CtIP initiates DNA end resection at the DSB, creating a 3′ protruding single-stranded DNA (ssDNA) tail. The ssDNA strand is then coated with recombinase RAD51 in a BRCA2-dependent manner. Then, the resulting ssDNA-RAD51 filament invades the donor DNA (strand invasion) and anneals to complementary homologous sequences. Finally, the defective DNA sequence is newly synthesized from the 3′ ssDNA end, using the donor DNA as a template (Goodarzi and Jeggo 2013). When exogenous DNA is used as a donor, a new DNA sequence is incorporated at the Cas9 cleavage site if it is engineered between the two homology arms. Therefore, a DNA sequence adjacent to the Cas9-induced DSB can be edited as desired. However, HR is not a dominant repair pathway in human somatic cells. Therefore, the efficiency of donor DNA incorporation at the DSB site is still poor in these cells.
The predominant DSB repair pathway is NHEJ, which directly joins the two DSB ends. This pathway is usually precise but sometimes mutagenic (Bétermier et al. 2014). If the DSB ends fuse and restore the wild-type sequence, then the restored DNA sequence is retargeted and recleaved by Cas9. The process of cleaving and joining the cleaved ends continues until mutagenic NHEJ creates an insertion or deletion (indel) at the cut site. Therefore, target genes frequently contain indels, even if one of the two autosomal alleles is successfully edited as intended by HR. Furthermore, off-target DSB cleavage can also lead to indel mutations. To reduce indels at off-target sites, double nicking of opposite strands by the RuvC dead mutant Cas9D10A with two gRNAs has been proposed (Ran et al. 2013a; Shen et al. 2014). When two targets are designed in close proximity, the double nick creates a DSB. Because most off-target nicks are not adjacent to each other and do not form DSBs, these nicks are repaired precisely and rarely lead to indels (Ran et al. 2013a; Shen et al. 2014). However, this strategy cannot avoid the creation of indels at on-target sites. A nick at the target site triggers HR without a DSB, but at a low efficiency (Davis and Maizels 2014; Vriend et al. 2016).
In this study, we developed a precise and efficient nucleotide substitution technology that does not (1) generate a DSB that leads to indels; (2) necessitate drug selection requiring integration of a drug-resistant gene into the genome; or (3) disequilibrate the balance of DNA repair pathways, either through NHEJ inhibitors, HDR enhancers, or silencing of DNA repair genes, all of which may cause global genomic instability.
Results
A tandem nick (TN) in the genome induces efficient gene editing using a plasmid donor
We devised an assay system that enabled simple and accurate assessment of single-nucleotide substitution efficiency. A single copy of the mCherry-P2A-EGFP reporter gene with a c.321C>G: p.Y107* EGFP mutation (EGFPcC>G reporter) was integrated into the genome of 293T cells (Fig. 1A). Cells expressed mCherry, but the EGFP expression was disrupted by the nonsense mutation. EGFP expression was restored when c.321G was reverted to C by nucleotide substitution. The extent of gene correction can be monitored by EGFP expression through flow cytometric analyses (Fig. 1A).
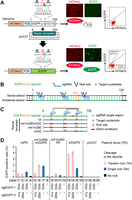
Nucleotide substitution by tandem nick (TN) of the reporter gene. (A) Schematic of the nucleotide substitution reporter assay. Representative data from flow cytometry and epifluorescent images of 293T EGFPcC>G reporter cells are also presented. (B) Diagram of the EGFPcC>G reporter and sgRNAs. (C) Diagram of the repair templates. (D) Nucleotide substitution efficiency as measured by flow cytometry (mean ± SD, N = 3) is presented. The indicated sgRNAs were expressed with Cas9D10A. The indicated repair templates were introduced into the cells as plasmid donors. Also see Supplemental Figure S1.
We designed three EGFP-specific gRNA sequences that were complementary to the sense strand DNA sequence of the EGFP gene (Fig. 1B) and introduced a site-specific nick in the sense strand of the EGFPcC>G reporter by transfecting reporter cells with PX462 plasmids (Ran et al. 2013b) that coexpressed Cas9D10A and EGFP-specific sgRNA (sgEGFP). We also transfected reporter cells with a donor plasmid, which was constructed by inserting a DNA sequence homologous to the EGFP gene sequence from 4 to 720 bp (the EGFP repair template) into the pUC57 plasmid (Fig. 1A). We analyzed a proportion of EGFP-positive cells after the gene-editing process and following cell culture without the enrichment of plasmid-transfected cells by drug selection.
It is well-known that a single nick (SN) does not efficiently induce gene editing (Ran et al. 2013a; Davis and Maizels 2014). Therefore, we investigated whether two nicks in the same strand of the EGFPcC>G reporter could promote nucleotide substitution. We first introduced a TN in the EGFPcC>G reporter by nicking either sgEGFP41s and sgEGFP332s sites or sgEGFP260s and sgEGFP332s sites (Fig. 1C). When a donor plasmid harboring a wild-type EGFP gene sequence (wtPD) (Fig. 1C) was used, SNs at the sgEGFP332s site in both the EGFPcC>G reporter and donor plasmid achieved up to 1% nucleotide substitution (Fig. 1D, lane 3; Supplemental Fig. S1). The TNs in both the reporter gene and wtPD led to less efficient nucleotide substitution than the SNs (Fig. 1D, lanes 1,2; Supplemental Fig. S1). These data suggested that nicks in the repair template might negatively affect gene editing. Therefore, we repeated the assay using a repair template containing three silent mutations in all three sgEGFP target regions (target region: target sequence + PAM sequence) to prevent the donor plasmid from being nicked (m41m260m332PD) (Fig. 1C). m41m260m332PD improved the efficiency of SN-induced nucleotide substitution by about twofold when compared with wtPD (Fig. 1D, cf. lanes 3,7). However, the efficiency of TN-induced nucleotide substitution using m41m260m332PD was either lower or the same as that of the SN-induced nucleotide substitution (Fig. 1D, lanes 5–7; Supplemental Fig. S1). Next, we constructed two more donor plasmids that were single-nicked while the EGFPcC>G reporter was nicked in tandem: m41m260PD contained three silent mutations in both the sgEGFP41s and sgEGFP260s target regions and could be nicked only at the sgEGFP332s site; m332PD contained three silent mutations in the sgEGFP332s target region and could be nicked only at the sgEGFP41s or sgEGFP260s site (Fig. 1C). The TNs in the EGFPcC>G reporter did not cause nucleotide substitution when m41m260PD was used as a donor (Fig. 1D, lanes 9–12; Supplemental Fig. S1). In contrast, the TNs generated approximately 4% EGFP-positive cells when m332PD was used as a donor (Fig. 1D, lanes 13–16; Supplemental Fig. S1).
We subsequently introduced a TN in the EGFPcC>G reporter, by nicking either the sgEGFP332s and sgEGFP504s sites or the sgEGFP332s and sgEGFP612s sites (Supplemental Fig. S2), and tested whether they promoted efficient nucleotide substitution using m332PD as a donor. These TNs also induced efficient nucleotide substitution, although the target nucleotide was not located between the two nicks (Supplemental Fig. S2C, lanes 2,3). These data suggest that a TN at the sgEGFP332s site and at another site within the EGFPcC>G reporter promotes gene editing, as long as the donor plasmid is single-nicked at any site in the repair template, excluding the sgEGFP332s site (Supplemental Fig. S3A–C).
SNs in the genome and donor plasmid facilitate efficient nucleotide substitution
The tandem-nicking experiments indicated that the distance between the two nicks in the target gene did not affect the gene-editing efficiency when nicked plasmids were used as repair templates (Fig. 1D, lanes 13,14; Supplemental Fig. S2C, lanes 2,3; Supplemental Fig. S3B). Based on these results, we hypothesized that an SN at the sgEGFP332s site of the EGFPcC>G reporter was sufficient to induce gene editing when the donor plasmid was nicked once. To test this, we investigated whether a SN in the backbone of the donor plasmid can stimulate gene editing if the EGFPcC>G reporter is single-nicked at the sgEGFP332s site (Supplemental Fig. S3D). We designed seven pUC57-specific sgRNAs (sgUC57s) (Fig. 2A). These sgRNAs and sgEGFP41s (the latter used as a positive control) targeted the same strand of the donor plasmid. As expected, all the sgUC57s in combination with sgEGFP332s led to efficient nucleotide substitution (Fig. 2B, lanes 13–19). The efficiency of nucleotide substitution by a combination of SNs in the target gene and donor plasmid (SNGD) was equal to or greater than that by a TN (Fig. 2B, cf. lane 12 and lanes 13–19).
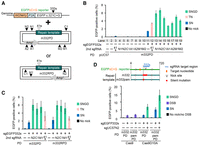
Nucleotide substitution by a combination of single nicks in the reporter gene and backbone of the donor plasmid (SNGD). (A) Schematic of the nucleotide substitution reporter assay by SNGD. sgRNA target sites are indicated by triangles. (B,C) Nucleotide substitution efficiency as measured by flow cytometry (mean ± SD, N = 3 [B] or N = 4 [C]) is presented in each panel. A nick in the EGFPcC>G reporter was introduced at the sgEGFP332s site. A nick in the backbone of the donor plasmid was introduced at one of the sgUC57 sites. m332PD (B) or m332RPD (C) was used as the donor. (D) Efficiency of the SNGD-mediated or DSB-mediated nucleotide substitution as measured by flow cytometry (mean ± SD, N = 3) is presented.
Next, we used a donor plasmid in which the EGFP repair template had been inserted into pUC57 in reverse orientation to m332PD (m332RPD). In this assay, pUC57s and sgEGFP41s targeted different strands of m332RPD (Fig. 2A). sgUC57N2, sgUC57C1, and sgUC57M1 also led to efficient gene editing when m332RPD was used as a donor (Fig. 2C). These data indicated that a single nick in either strand of the donor plasmid could stimulate SNGD-mediated gene editing (Supplemental Fig. S3D).
To prevent Cas9D10A from nicking donor plasmids at the sgEGFP332s site, we designed m332PD to contain three silent mutations in the sgEGFP332s target region. However, minimizing the number of silent mutations in the successfully edited gene is preferable, because such mutations may affect RNA stability, splicing, and gene translation, although they do not alter the amino acid sequence of proteins (Gingold and Pilpel 2011). Therefore, we performed a nucleotide substitution assay using a donor plasmid with a single silent mutation in the PAM sequence of the sgEGFP332s target region (m332pamPD). We achieved 14.2% nucleotide substitution when we introduced SNGD at the sgEGFP332s and sgUC57N2 sites (Fig. 2D). In contrast, nucleotide substitution efficiency was much lower (2.0%) when we introduced a DSB to the EGFPcC>G reporter at the sgEGFP332s site by transfecting with PX459-sgEGFP332s that coexpressed wild-type Cas9 and sgEGFP332s (Fig. 2D). Similarly, the SNGD method achieved a higher nucleotide substitution efficiency than the DSB method in HeLa EGFPcC>G reporter cells (Supplemental Fig. S4A).
We considered whether a nick not only at the sgEGFP332s site but also at other sites in the EGFPcC>G reporter can induce SNGD-mediated gene editing. To investigate this, we designed three new sgEGFPs that induce nicks in the sense strand of the EGFPcC>G reporter with Cas9D10A (Supplemental Fig. S4B). SNGD using one of these sgEGFPs and one of sgUC57s showed more efficient nucleotide substitution than an SN using identical sgEGFPs (Supplemental Text; Supplemental Fig. S4C–E). However, the efficiency of nucleotide substitution by SNGD using any of these sgEGFPs was lower than that using sgEGFP332s, suggesting that the efficiency of SNGD differs among sites. Next, we performed SNGD-mediated gene editing by nicking the anti-sense strand of the EGFPcC>G reporter. However, it did not lead to efficient nucleotide substitution (Supplemental Text; Supplemental Fig. S3B,F–K). These data suggest that nicking the sense strand of the target gene is favorable for efficient SNGD-mediated gene editing.
Reporter gene editing by SNGD is more precise than that by a Cas9-induced DSB
To directly compare the accuracy of gene editing by SNGD with a Cas9-induced DSB, we created another 293T reporter cell line, wherein we integrated two independent reporters into the genome: one copy of mCherry-P2A-EGFP c.321C>G and one copy of tagBFP-P2A-EGFP c.321C>G (Fig. 3A). We introduced a nick or a DSB at the sgEGFP332s target site of each reporter gene. We also introduced a nick at the sgUC57N2 site of m332pamPD for SNGD. Thereafter, we sorted EGFP-positive cells into 96-well plates (one cell/well) and cultured them to establish single-cell-derived clones. We analyzed the DNA sequences of both reporter genes in each single-cell clone (Fig. 3A).
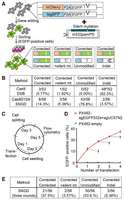
Sequence analysis of the reporter gene after the SNGD-mediated gene editing. (A) Diagram of the nucleotide substitution assay using the bi-EGFPcC>G reporter cells. (*) c.321C>G mutation. (B) Summary of the sequence analysis of the EGFPcC>G reporter in the EGFP-positive bi-EGFPcC>G reporter single-cell clones. The clones were classified into four types based on the sequencing results: (1) Both reporters were corrected as designed; (2) one copy was corrected and the other incorporated the silent mutation of the repair template, but the c.321C>G mutation was not corrected; (3) one copy was corrected and the other was not modified; and (4) one copy was corrected and the other resulted in indel (also see A): (#) One clone contained two nucleotide substitutions near the DSB site without an indel. (C–E) The SNGD process was repeated. (C) The schedule of the SNGD cycle is shown. (D) Gene-editing efficiency as measured by flow cytometry is shown (mean ± SD, N = 3). (Solid line) SNGD; (dashed line) no nick. (E) A summary of the sequence analysis of the EGFPcC>G reporter in the EGFP-positive bi-EGFPcC>G reporter single-cell clones after three consecutive rounds of the SNGD process.
When wild-type Cas9 was used for gene editing, we detected indels or mutations in the reporter genes in 92.3% of EGFP-positive single-cell clones that contained at least one copy of the correctly edited reporter gene. In contrast, when gene editing was performed using SNGD, indels were detected in only 3.57% of EGFP-positive single-cell clones (Fig. 3B). SNGD successfully corrected both copies of the reporter at a higher efficiency (14.3% of EGFP-positive cells) than a DSB (5.77% of EGFP-positive cells) (Fig. 3B). The high unmodified copy frequency in the EGFP-positive cells suggested that the sgEGFP332s target region remained intact in the EGFP-negative cells. If true, repeating the SNGD process could achieve a higher nucleotide substitution efficiency. Indeed, the rate of EGFP-positive cells reached 34% after three consecutive rounds of the SNGD process (Fig. 3C,D). We also analyzed the EGFPcC>G reporter sequence of EGFP-positive single-cell subclones. Both copies of the reporter were correctly edited in 21 of 56 (37.5%) EGFP-positive single-cell clones, whereas only three of the 56 clones (5.36%) possessed indels (Fig. 3E). The fourth round of SNGD further increased the EGFP-positive fraction to 41% (Fig. 3D). These data indicate that SNGD achieves efficient and precise correction of the mutated EGFP gene by nucleotide substitution.
The target gene incorporates a long DNA sequence from the repair template through SNGD-mediated gene editing
To obtain mechanistic insights into how the reporter gene incorporates the repair template sequence, we investigated the effect of the length of the repair template on the SNGD-mediated gene editing. We inserted different truncated sequences of m332pam EGFP repair templates into pUC57 and used these plasmids as donors (Fig. 4A). Consequently, longer EGFP repair templates induced more efficient SNGD-mediated nucleotide substitution in 293T and HeLa EGFPcC>G reporter cells (Fig. 4B,C). Then, we investigated the extent to which the EGFP repair template of the donor plasmid was incorporated into the EGFPcC>G reporter gene through gene correction. We performed SNGD-mediated gene editing using a donor plasmid containing four or 17 additional silent mutations that spanned the entire repair template of m332pamPD (m332pamTrick4PD/Trick17PD) (Fig. 4D). The gene-editing efficiency using these donors was not as high as that of m332pamPD (Fig. 4E), suggesting that reducing the homology between the EGFPcC>G reporter and donor sequence suppressed SNGD-mediated gene editing. After the SNGD process, we analyzed the DNA sequence of the EGFPcC>G reporter of each EGFP-positive single-cell clone. When we used m332pamTrick17PD, >67% of EGFP-positive 293T single-cell clones and >48% of EGFP-positive HeLa single-cell clones incorporated DNA sequences longer than 100 bp from the repair template (Supplemental Fig. S5A,B). It is possible that many of the mismatches between the genome and repair template induced long-repair template incorporation by the genome. However, even if we use m332pamTrick4PD, >66% of EGFP-positive 293T single-cell clones incorporated DNA sequences longer than 178 bp from the repair template (Supplemental Fig. S5C). These data suggest that SNGD uses HR-like homology-directed repair for gene editing.
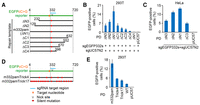
Efficiency of SNGD-mediated gene editing using various types of repair templates. (A,D) Diagrams of the repair templates. (B,C,E) Nucleotide substitution efficiency as measured by flow cytometry (mean ± SD, N = 3) is presented in each panel. The EGFPcC>G reporter was nicked at the sgEGFP332s site, and donor plasmids were nicked at the sgUC57N2 site. The indicated repair template was used as a plasmid donor in each assay. 293T EGFPcC>G reporter cells were used in B and E. HeLa EGFPcC>G reporter cells were used in C.
Noncanonical HDR promotes SNGD-mediated gene editing
We subsequently investigated whether known HR factors are involved in the Cas9-mediated gene editing process. We knocked down the major protein factors required for the canonical HR in HeLa EGFPcC>G reporter cells using siRNA (Supplemental Fig. S6A–D). We previously confirmed suppression of HR by siRNA-mediated CtIP knockdown using a direct repeat GFP (DR-GFP) assay (Pierce et al. 1999; Kato et al. 2014). We also confirmed abrogation of HR by siRNA-mediated knockdown of BRCA2 or RAD51 by DR-GFP assay (Supplemental Fig. S5E). Depletion of CtIP, BRCA2, or RAD51 suppressed DSB-induced nucleotide substitution (Fig. 5A,B). Knockdown of BRCA2 or RAD51 markedly suppressed the nucleotide substitution by SN (Fig. 5C). However, CtIP depletion did not inhibit nucleotide substitution by SN (Fig. 5D). These data suggest that an SN in the genome can stimulate RAD51-dependent recombination between the genome and donor plasmids without CtIP-dependent DNA end resection. SNGD also promoted nucleotide substitution in the absence of CtIP, being as efficient as in the presence of CtIP (Fig. 5E). In contrast to SN-mediated gene editing, SNGD facilitated efficient nucleotide substitution in RAD51-depleted cells (Fig. 5F). Although RAD51 was dispensable, SNGD-mediated gene editing partially depended on BRCA2 (Fig. 5F), suggesting the BRCA2's support of SNGD-mediated gene editing via mechanisms other than promoting the formation of RAD51 nucleofilaments on ssDNA. RAD52 is required for HDR via single-strand annealing (Stark et al. 2004). In addition, RAD52 promotes HR in the absence of BRCA2 (Feng et al. 2011). Therefore, we investigated whether RAD52 depletion suppresses SNGD-mediated gene editing in both RAD52-BRCA2 double knockdown and RAD52 single knockdown cells (Supplemental Fig. S6F). RAD52 knockdown did not suppress SNGD-mediated gene editing in BRCA2-depleted or BRCA2-positive cells (Fig. 5G). Rather, RAD52 single knockdown stimulated more efficient SNGD-mediated nucleotide substitution. Thus, SNGD utilizes noncanonical HDR for recombination between the genome and donor plasmid.
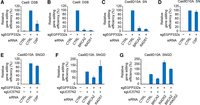
Gene-editing efficiency in HDR factor-depleted cells. The relative gene correction efficiency in the indicated siRNA-transfected cells (relative to the gene-editing efficiency in siCTRL-transfected cells; mean ± SD, N = 3) is presented in each panel. m332pamPD was used as a donor for all experiments. (A,B) DSB-mediated gene editing in CtIP-, BRCA2-, or RAD51-depleted HeLa EGFPcC>G reporter cells. A DSB was introduced at the sgEGFP332s target site of the reporter. (C,D) SN-mediated gene editing in BRCA2-, RAD51-, or CtIP-depleted HeLa EGFPcC>G reporter cells. A nick was introduced at the sgEGFP332s target site of the reporter. (E,F) SNGD-mediated gene editing in CtIP-, BRCA2-, or RAD51-depleted EGFPcC>G reporter cells. (G) SNGD-mediated gene editing in RAD52- and/or BRCA2-depleted HeLa EGFPcC>G reporter cells. Nicks were introduced at the sgEGFP332s target site of the reporter and at the sgUC57N2 site of the donor plasmid for SNGD.
SNGD can edit endogenous loci
We applied the SNGD method to edit endogenous loci. We designed gene-editing experiments to create a restriction enzyme BamHI recognition site at the EMX1.13 site (Hsu et al. 2013) and a XhoI recognition site near HEK293 site 3 (Tsai et al. 2015) by single-nucleotide substitutions (Supplemental Fig. S7A). Each site-specific sgRNA targeted the DNA sequence to introduce a nick/DSB at 20 bp (EMX1 locus) or 30 bp (HEK293 locus) downstream from the target nucleotide. Each repair template contained a mutation in the PAM sequence and BamHI (for EMX1 locus) or XhoI (for HEK293 locus) recognition sequence. We used PX461/PX458 plasmids that coexpressed Cas9D10A/Cas9-P2A-GFP and site-specific sgRNA. We obtained cells that had been successfully transfected with the plasmids by sorting GFP-positive cells. The target loci in the sorted cells were amplified by PCR for restriction fragment length polymorphism (RFLP) analysis using the BamHI or XhoI enzyme. The cleaved DNA fragments indicated successful gene editing. As shown in Supplemental Figure S7B, the SNGD method successfully edited both target loci. We further confirmed gene editing by PCR using high single-nucleotide discrimination (HiDi) DNA polymerase that efficiently discriminates primers with a mismatch at the 3′ end (Drum et al. 2014). The edited alleles, but not non-edited alleles, can be amplified by HiDi PCR using the edited allele-specific PCR primer. As shown in Supplemental Figure S7C, SNGD successfully edited the EMX1 and HEK293 loci. SNGD showed gene-editing efficiency comparable to or better than that of a DSB (Supplemental Fig. S7C). Semiquantitative HiDi PCR showed that the SNGD-mediated gene editing efficiency was ∼4% (Supplemental Fig. S7D).
Correction of 1-bp insertion in the thymidine kinase 1 (TK1) gene by SNGD-mediated gene editing
Finally, we used the SNGD method to correct a mutated endogenous gene. TSCER2 cells are compound heterozygous for the TK1 gene; one allele contains a 1-bp insertion in exon 4 that induces a frameshift; the other contains a G-to-A transition in exon 5 that abrogates the TK1 function (Honma et al. 2007). Therefore, TSCER2 cells are sensitive to cytidine-hypoxanthine-aminopterin-thymidine (CHAT) medium (Honma et al. 2007). Successful correction of one of these mutations rendered cells resistant to CHAT medium. We targeted the 1-bp insertion in exon 4. We designed an sgRNA target sequence including the 1-bp insertion at its 3′ end (Fig. 6A). This sgRNA does not target the wild-type TK1 gene. Therefore, this experimental design facilitated the use of the repair template without any silent mutation. The sgRNA was coexpressed with Cas9D10A/Cas9-P2A-GFP in TSCER2 cells. We sorted GFP-positive cells, cultured the sorted cells in normal media for a week, and then cultured the cells in CHAT medium. In this experimental setting, 45% of cells became CHAT-medium resistant after DSB-mediated gene editing (Fig. 6B; Supplemental Fig. S8A). Although the SNGD method was less efficient than the conventional method, 9.6% of cells achieved CHAT-medium resistance after the SNGD-mediated gene editing (Fig. 6B; Supplemental Fig. S8A). It is considered that restoration of the reading frame by a mutagenic NHEJ-induced indel can also rescue the kinase activity of TK1 protein, although the gene-edited allele shows gene conversions, which can lead to the misinterpretation of gene correction. In order to rule this out, we sequenced exon 4 of the TK1 gene to confirm that the mutant allele had been correctly edited. All CHAT-medium-resistant clones (92/92) showed the correct TK1 gene sequence after the SNGD-mediated gene editing (Fig. 6C; Supplemental Fig. S8B). In contrast, 18 of 92 CHAT-medium-resistant clones showed an incorrect 1-bp deletion in the DSB-mediated gene-editing process (Fig. 6C; Supplemental Fig. S8B). Thus, SNGD could correct the mutated TK1 gene with marked accuracy and moderate efficiency.
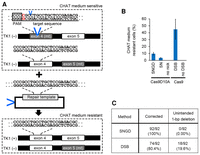
SNGD-mediated gene editing of a mutated thymidine kinase 1 (TK1) gene. (A) Schematic of correction of the TK1 gene in the TSCER2 cell line. TSCER2 cells possess both exon 4- and exon 5-mutated alleles and are sensitive to CHAT medium. The single-nucleotide insertion in exon 4 (indicated in red) was targeted to correct the TK1 gene. When the mutation in exon 4 was edited to recover TK activity, the cells acquired CHAT-medium resistance. The PAM sequence is indicated by a box. The sgRNA target sequence is underlined. Nick sites in the TK1 gene and donor plasmid are indicated by the blue V mark. (B) Percentages of CHAT-medium-resistant cells are indicated (mean ± SD, N = 3). (C) Summary of sequence analysis of exon 4 of the TK1 gene in CHAT-medium-resistant cell clones.
Discussion
In this study, using nicked plasmid donors, we demonstrated that an SN in the target gene promotes efficient gene editing with markedly lower levels of indels. In a previous study, using nickase-type homing nucleases, it was shown that nicking a repair template of the plasmid donor improves nick-induced HDR efficiency (Davis and Maizels 2014). Although homing nucleases recognize rare and specific DNA sequences, Cas9 can recognize various DNA sequences with gRNAs. This flexible characteristic of Cas9 enables us to utilize SNGD-mediated HDR for practical gene editing. Furthermore, because any repair templates can be cloned into pUC57 plasmids, we can use sgUC57s to edit any genes by SNGD. Thus, our study has revealed a precise and efficient gene-editing strategy without any special requirements.
DSB-mediated gene editing depends on CtIP, BRCA2, and RAD51, i.e., canonical HR (Fig. 5A,B; Supplemental Fig. S9A-i). In contrast, SN-mediated gene editing depends on RAD51 but not CtIP (Fig. 5C,D), suggesting that a 3′-ended ssDNA tail is generated at a Cas9D10A-created nick without DNA end resection (Supplemental Fig. S9A-ii). As a previous study proposed a model whereby DNA nicks are unwound (Davis and Maizels 2014, 2016), the ssDNA tail may be created by some helicases. As another possibility, we propose that transcription and R-loop formation may contribute to the unwinding of the DNA double helix (Supplemental Fig. S9B). R-loops comprise nascent RNA hybridized to template DNA and single-stranded nontemplate DNA. R-loops are preferentially formed when the nontemplate strand is G-rich (Aguilera and García-Muse 2012; Skourti-Stathaki and Proudfoot 2014) or nicked (Roy et al. 2010). From this aspect, a Cas9D10A-induced nick in the sense strand may be a preferable R-loop initiation zone. This model is consistent with a previous report that transcription stimulates nick-induced HDR with single-stranded deoxyoligonucleotide (SSO) (Davis and Maizels 2014). After the 3′-ended ssDNA is exposed, RAD51 coats the ssDNA and promotes recombination.
Although SN-induced gene editing requires RAD51, SNGD enables RAD51- and RAD52-independent gene editing using a nicked donor plasmid for the repair template (Fig. 5F,G). These data suggest that strand invasion is no longer required for recombination between the nicked genome and nicked donor plasmid. Although clarifying the underlying molecular mechanisms will require further investigation, we consider that Cas9D10A induces structural changes in the donor plasmid (Supplemental Fig. S9A-iii). These changes facilitate high-level accessibility between the nicked genome and donor plasmids, enabling the single-stranded 3′ DNA tails in the genome to anneal to the complementary DNA sequence in the donor plasmid without RAD51-dependent strand invasion (Supplemental Fig. S9A-iii). Since an ssDNA tail at a nick anneals to a complementary SSO and promotes annealing-driven strand synthesis in a RAD51-independent manner (Davis and Maizels 2016), we can speculate that the nicked donor plasmid may be single-stranded and utilized as a ssDNA repair template during SNGD-mediated gene editing (Supplemental Fig. S9A-iii). Davis and Maizels (2016) also propose another RAD51-independent repair pathway: When SSO donors that are complementary to the intact target strand of the target gene are provided, the SSO anneals to the intact strand at the nick. The latter pathway may also promote SNGD-mediated gene editing (Supplemental Fig. S9A-iii).
Two CRISPR-based gene correction strategies were recently reported: The first does not require DSB, and the second relies on DSB repair. The former approach fuses catalytically inactive Cas9 or Cas9D10A with the enzyme cytidine deaminase. However, this approach can only induce C:G to T:A transitions (Komor et al. 2016; Nishida et al. 2016), whereas our approach can correct any combination of nucleotide conversions. Furthermore, our SNGD-mediated gene editing could successfully correct the 1-bp insertion (Fig. 6), but it cannot be corrected with the former approach. The second approach, homology-independent targeted integration (HITI), is advantageous for inserting a DNA sequence long enough to form a circular shape into nondividing cells. However, duplicates of ∼6 and ∼17 nt occur at two junctions after HITI-mediated gene editing (Suzuki et al. 2016). Furthermore, this method relies on DSB generation using wild-type Cas9; therefore, it is susceptible to off-target cleavage effects as well as higher numbers of indels. Therefore, HITI is not suitable for precise nucleotide substitution; however, SNGD offers a superior method as it overcomes the problems associated with these recently developed approaches.
For ex vivo gene therapies, there are two options: (1) perform the gene correction procedure on cells, select one precisely gene edited cell clone, expand in vitro, and transplant, which is safer but very expensive; and (2) perform the gene correction procedure on cells and then transplant without selection, which is cost-effective. If the gene-corrected cells have a growth advantage compared with diseased cells, the second method is practical as gene therapy to treat severe diseases; for example, primary severe combined immunodeficiency can be treated with the second option (Fischer et al. 2001). The precise gene-editing property of SNGD would make the second option advantageous and safer.
Methods
Plasmids, DNA oligos, siRNAs, cells, and antibodies
The DNA sequences of mCherry-P2A-EGFP c.321C>G and tagBFP-P2A-EGFP c.321C>G are presented in Supplemental Table S1. Cas9-sgRNA coexpression plasmids (pSpCas9 plasmids; gifts from Dr. F. Zhang) are shown in Supplemental Table S2. Target sequences are listed in Supplemental Table S3. Repair templates are listed in Supplemental Tables S4, S5. Oligo DNA primers for PCR or Sanger sequencing are presented in Supplemental Table S6. siRNA sequences are listed in Supplemental Table S7. pCBASceI was a gift from Dr. M. Jasin. Details regarding EGFPcC>G reporter cells and antibodies are provided in Supplemental Methods.
Nucleotide substitution reporter assay
Transfection of 293T or HeLa cells with plasmids was performed using Effectene Transfection Reagent (Qiagen) or the Neon Transfection System (Invitrogen), respectively. Transfection of HeLa cells with siRNAs was performed using Lipofectamine RNAiMAX (Invitrogen). Four (293T) or five (HeLa) days after transfection with pSpCas9 and donor plasmids, the cells were subjected to flow cytometry using FACSCalibur (BD). Details are provided in Supplemental Methods.
Sequence analysis of the EGFPcC>G reporter gene
Single cells expressing EGFP were plated into a 96-well tissue culture plate using a cell sorter FACSAria IIu (BD). The cells were cultured for 2 wk. Genomic DNA of the single-cell clones was extracted using the MonoFasII cell DNA extraction kit (GLScience) according to the manufacturer's protocol. DNA fragments containing the EGFP sequence were PCR-amplified using KOD Plus Neo (Toyobo) with the primer sets shown in Supplemental Table S6. The DNA sequences of the PCR products were analyzed by the Sanger method (Fasmac or Eurofins Genomics).
TK1 gene correction
For SNGD-mediated gene editing, PX461-sgTK(ex4) and PX462-sgUC57N2 transfections were performed. For SN-mediated gene editing, PX461-sgTK(ex4) and PX462-empty transfections were performed. For DSB-mediated gene editing, PX458-sgTK(ex4), PX459-empty, and donor plasmids were used for transfection. The transfected cells were incubated in the culture medium for 2 d. Then, GFP-positive cells were sorted by FACSAria IIu. After incubation in normal culture medium for 5 d, cells were seeded in a total volume of 200 µL of CHAT medium (10 µM 2′-deoxycytidine [Sigma], 200 µM hypoxanthine [Sigma], 100 nM aminopterin [Sigma], and 17.5 µM thymidine [Sigma]) at a density of 1.25, 5, or 20 cells/well in two 96-well plates. Two weeks after incubation in CHAT medium, numbers of colony-positive wells were analyzed. For DNA sequence analysis, cell colonies were directly subjected to PCR using MightyAmp DNA Polymerase ver. 2 (Takara), and the PCR products underwent DNA sequence analyses (Fasmac). Primers for PCR and DNA sequencing are listed in Supplemental Table S6. Details are provided in Supplemental Methods.
Data access
All sequencing data from this study have been submitted to the DNA Data Bank of Japan (DDBJ; http://www.ddbj.nig.ac.jp/index-e.html) under accession numbers LC334156–LC334339 and LC336811–LC337230.
Acknowledgments
We thank F. Zhang and M. Jasin for plasmids. We also thank T. Takahashi, T. Ogi, B. Shiotani, T. Mashimo, M. Ohtsuka, and H. Sasanuma for useful discussions, and K. Nanba for technical support. We also thank CentMeRE, Graduate School of Medicine, Osaka University, for technical assistance. This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (JP26241014 and JP16K12596: S.N.), the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (17ek0109229s07 and 16ek0109035h0003: S.N.), a Grant-in-Aid for JSPS Research Fellow (JPA15J047380: K.N.), the Takeda Science Foundation, The Naito Foundation, and The Sumitomo Foundation (S.N.).
Author contributions: Conceptualization and methodology are attributed to S.N.; K.N., Y.Z., A.T., Y.H., and S.N. performed the investigation; S.N. wrote the paper; and S.N. and C.B.G. reviewed and edited the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226027.117.
- Received June 7, 2017.
- Accepted November 27, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








