
Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis
- Hector L. Franco1,6,
- Anusha Nagari1,
- Venkat S. Malladi1,
- Wenqian Li2,
- Yuanxin Xi3,
- Dana Richardson4,
- Kendra L. Allton5,
- Kaori Tanaka5,
- Jing Li5,
- Shino Murakami1,
- Khandan Keyomarsi4,
- Mark T. Bedford2,
- Xiaobing Shi5,
- Wei Li3,
- Michelle C. Barton5,
- Sharon Y.R. Dent2 and
- W. Lee Kraus1
- 1Laboratory of Signaling and Gene Regulation, Cecil H. and Ida Green Center for Reproductive Biology Sciences and Division of Basic Reproductive Biology Research, Department of Obstetrics and Gynecology, University of Texas Southwestern Medical Center, Dallas, Texas 75390, USA;
- 2Department of Epigenetics and Molecular Carcinogenesis and The Center for Cancer Epigenetics, University of Texas MD Anderson Cancer Center, Smithville, Texas 78957, USA;
- 3Department of Molecular and Cellular Biology and Division of Biostatistics, Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, Texas 77030, USA;
- 4Department of Experimental Radiation Oncology, University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 5Department of Epigenetics and Molecular Carcinogenesis, University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences and The Center for Cancer Epigenetics, University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA
Abstract
Noncoding transcription is a defining feature of active enhancers, linking transcription factor (TF) binding to the molecular mechanisms controlling gene expression. To determine the relationship between enhancer activity and biological outcomes in breast cancers, we profiled the transcriptomes (using GRO-seq and RNA-seq) and epigenomes (using ChIP-seq) of 11 different human breast cancer cell lines representing five major molecular subtypes of breast cancer, as well as two immortalized (“normal”) human breast cell lines. In addition, we developed a robust and unbiased computational pipeline that simultaneously identifies putative subtype-specific enhancers and their cognate TFs by integrating the magnitude of enhancer transcription, TF mRNA expression levels, TF motif P-values, and enrichment of H3K4me1 and H3K27ac. When applied across the 13 different cell lines noted above, the Total Functional Score of Enhancer Elements (TFSEE) identified key breast cancer subtype-specific TFs that act at transcribed enhancers to dictate gene expression patterns determining growth outcomes, including Forkhead TFs, FOSL1, and PLAG1. FOSL1, a Fos family TF, (1) is highly enriched at the enhancers of triple negative breast cancer (TNBC) cells, (2) acts as a key regulator of the proliferation and viability of TNBC cells, but not Luminal A cells, and (3) is associated with a poor prognosis in TNBC breast cancer patients. Taken together, our results validate our enhancer identification pipeline and reveal that enhancers transcribed in breast cancer cells direct critical gene regulatory networks that promote pathogenesis.
Gene expression profiling has shown that breast cancer is not a single disease with variable morphologic features and biomarkers, but rather a group of molecularly distinct neoplastic disorders (Perou et al. 2000; Sotiriou and Pusztai 2009; Ciriello et al. 2015). These analyses have identified at least five distinct molecular subtypes of breast cancer—Luminal A, Luminal B, HER2+, Triple negative (TN)/Basal-like, Triple negative/Claudin-low—which correlate with hormone responsiveness, patient prognosis, and response to therapy and that express unique sets of genes that persist from preneoplastic lesions through metastatic disease (Perou et al. 2000; Sotiriou and Pusztai 2009). Nonetheless, little is known about the initial, disease-driving transcriptional and epigenetic changes that promote the phenotypic outcomes.
Deep sequencing technologies used to interrogate the genome have pointed to transcriptional enhancers, as well as the transcription factors (TFs) that promote their formation, as key regulatory elements controlling the cell-type–specific biology of essentially all biological systems examined to date (Shlyueva et al. 2014; Heinz et al. 2015). In cancers, cell-type–specific enhancers can become deregulated or “hijacked,” allowing the activation of genes that promote tumor formation and metastasis (Chipumuro et al. 2014; Northcott et al. 2014; Franco and Kraus 2015). Key questions regarding enhancer biology in cancers and other biological systems include (1) the location of enhancers throughout the genome, (2) the functionality or activity of the enhancers, (3) the TFs that nucleate enhancer formation, and (4) the target genes of the enhancers (Shlyueva et al. 2014; Heinz et al. 2015).
Although different enhancer-nucleating TFs may be expressed in different cell types, the enhancers that they form share several common features. For example, enhancers are typically found in open regions of chromatin (as assessed by DNase-seq) (Crawford et al. 2006; Sheffield et al. 2013) and are enriched with a common set of histone modifications (as assessed by ChIP-seq), including histone H3 lysine 4 monomethylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac) (Heintzman et al. 2007, 2009). Recently, genomic assays revealed that many enhancers are bound by RNA polymerase II (Pol II) and are actively transcribed, producing enhancer RNAs (eRNAs) (De Santa et al. 2010; Kim et al. 2010; Hah et al. 2011), which serve as a robust mark of active enhancers that can be used to track enhancer activity (Wang et al. 2011; Hah et al. 2013; Li et al. 2013; Franco et al. 2015; Kim and Shiekhattar 2015). We and others have shown that global run-on sequencing (GRO-seq) and its derivatives (e.g., PRO-seq) are robust genomic methods to detect and measure ongoing transcription at enhancers, which can be used for enhancer prediction (Hah et al. 2011, 2013; Wang et al. 2011; Core et al. 2014; Chae et al. 2015; Heinz et al. 2015).
In the studies presented herein, we describe the development of a computational pipeline, called the Total Functional Score of Enhancer Elements (TFSEE), which we used to mine a large number of transcriptomic and epigenomic data sets from human breast cancer cell lines, with the goal of identifying subtype-specific TFs that drive the subtype-specific biology of breast cancers.
Results
Generating transcriptional and epigenomic maps from breast cancer cells
To better understand the TF-driven transcriptional programs that control subtype-specific gene expression programs in breast cancers, we generated transcriptional and epigenomic maps for 13 different breast cell lines, 11 of which represent five intrinsic molecular subtypes of breast cancer (Fig. 1A), under basal growth conditions (i.e., without treatment). For these analyses, we used GRO-seq and RNA-seq, as well as ChIP-seq for 11 different histone modifications (Fig. 1A,B), the latter which is described in detail elsewhere (Y Xi, W Li, K Tanaka, KL Allton, D Richardson, J Li, HL Franco, A Nagari, V Malladi, LD Coletta, et al., in prep.). The histone modifications examined in this study are enriched at various functional elements and in a variety of chromatin environments, such as enhancers, promoters, gene bodies, and repressive regions of the genome, thus providing a broad survey of the epigenomic features associated with transcriptional outcomes (Fig. 1A,B; Supplemental Fig. S1A,B). In total, we generated data sets from 26 GRO-seq, 26 RNA-seq, and 286 ChIP-seq libraries (for details about replicates and read depth, see Supplemental Materials).
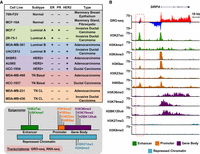
Transcriptional and epigenomic profiling identifies putative enhancers across the breast cancer genome. (A, top) Features of cell lines representing five distinct molecular subtypes of breast cancer, which were used in this study, including ER, PR, and HER2 status. (CL) Claudin-low. (Bottom) Depiction of the transcriptional (GRO-seq and RNA-seq) and epigenomic (ChIP-seq) profiles generated for each cell line. (B) Genome browser views of GRO-seq and histone modification ChIP-seq data from a normal breast epithelial cell line (76N-F2V) showing the features of a typical bidirectionally transcribed enhancer (red box with dashed line) and its nearest neighboring gene (SIRPA). Key features include transcription (red/blue), as well as histone modifications typically enriched at enhancers (green), promoters (brown), gene bodies (purple), and repressed chromatin (turquoise).
Although previous studies applied RNA-seq and ChIP-seq to transcriptomic and epigenomic analyses, respectively, in breast cancer cells, the inclusion of GRO-seq provides additional levels of unique information. GRO-seq is a direct measure of transcription, which yields the position and orientation of all actively transcribing RNA polymerases across the genome (Core et al. 2008), thus facilitating the comprehensive identification of transcribed regions and functional elements (Hah et al. 2011; Luo et al. 2014; Chae et al. 2015; Sun et al. 2015). Collectively, these data sets that we generated using these methods provide a unique resource for cancer researchers and provide a foundation for discovering the transcriptional and epigenomic underpinnings of breast cancer.
Using transcription to predict enhancers in the absence of other genomic information
We and others have shown that actively transcribed enhancers are more likely to (1) associate with enhancer-related chromatin modifications, such as H3K4me1 and H3K27ac, (2) loop to target gene promoters, and (3) correlate with target gene activation (Kim et al. 2010; Wang et al. 2011; Ørom and Shiekhattar 2013; Franco et al. 2015; Hah et al. 2015; Heinz et al. 2015). In addition, the so-called “super-enhancers” that are typically associated with oncogene expression are associated with enhancer transcription (Hah et al. 2015). Thus, enhancer transcription is a good predictor of active enhancers and can be used in the absence of other genomic information to predict active enhancers de novo (Hah et al. 2013; Chae et al. 2015).
We reasoned that using signatures of enhancer transcription from GRO-seq data would allow us to identify active enhancers in cell lines representing the different molecular subtypes of breast cancer that control the expression of cancer-relevant genes. We used a computational tool that we developed previously for calling unannotated transcription units from GRO-seq data, called groHMM (http://bioconductor.org/packages/release/bioc/html/groHMM.html) (Hah et al. 2011, 2013; Chae et al. 2015), to identify a set of eRNA transcripts produced in each of the 13 cell lines (Fig. 2A,B). This analysis revealed both unique and shared eRNAs across the cell lines (Fig. 2B), as well as an overlap of enhancer transcription with histone modifications that are typically enriched at enhancers (e.g., H3K27ac and H3K4me1) (Fig. 2C).
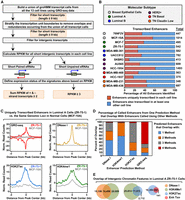
Unbiased, genome-wide prediction of active enhancers using GRO-seq data. (A) Overview of the computational pipeline used for the genome-wide annotation of enhancer transcripts (eRNAs) and prediction of active enhancers using GRO-seq data. (B) Catalog of all predicted active enhancers in the breast cancer cell lines listed in Figure 1A defined by enhancer transcription using the pipeline shown in A. Red indicates cell-type–specific enhancers, and blue represents enhancers transcribed in at least one other cell line. The colored circles indicate the molecular subtype of each cell line. (C) Metaplot analyses showing correlations among enhancer transcription, DNase I hypersensitivity, H3K27ac enrichment, and H3K4me1 enrichment for the uniquely transcribed enhancers identified in ZR-75-1 cells in B. Metaplots for the same genomic loci in MCF-10A cells are shown for comparison (gray lines). (D) Stacked bar chart comparing enhancer prediction methods in ZR-75-1 cells. Intergenic enhancers were called using prediction methods based on four different enhancer features: enhancer transcription (using GRO-seq and the pipeline shown in A), DNase I hypersensitivity, H3K4me1 enrichment, or H3K27ac enrichment. The percentage of called enhancers from each prediction method overlapping enhancers called using one or more of the other methods is shown. (E) Venn diagrams showing the number of enhancers called in ZR-75-1 cells and the overlap of enhancers called using DNase I and H3K4me1 (left), H3K4me1 and H3K27ac (middle), and H3K27ac and enhancer transcription (right).
Using the ZR-75-1 Luminal A cell line as an example (a cell line for which DNase I hypersensitivity data sets are publicly available), we compared the overlap of output from traditional enhancer prediction methods (e.g., DNase I hypersensitivity, or H3K4me1 and H3K27ac enrichment) (Shlyueva et al. 2014) to output from an enhancer transcription-based approach (Fig. 2D,E). Using the pipeline shown in Figure 2A, we found that 65% of enhancers called based on enhancer transcription using GRO-seq data are also identified by all three of the other methods (i.e., DNase I hypersensitivity, enrichment of H3K4me1 or H3K27ac). In contrast, only 1%–2% of enhancers called based on DNase I hypersensitivity, or enrichment of H3K4me1 or H3K27ac, are identified by all three of the other methods (Fig. 2D). This may be due, in part, to the fact that enhancer calling based on DNase I hypersensitivity, or H3K4me1 or H3K27ac enrichment, yields much larger numbers of putative enhancers (Fig. 2E), many of which may be false positives or inactive as true regulatory elements. Nonetheless, as we show below, incorporating enhancer transcription into an enhancer-calling pipeline that includes information about DNase I hypersensitivity, as well as H3K4me1 and H3K27ac enrichment, improves the fidelity of the enhancer calls.
Actively transcribed enhancers track with subtype-specific transcriptional programs
Next, we determined if the transcribed enhancers identified above might regulate nearby genes relevant to the biology of different subtypes of breast cancer. To do so, we identified all the uniquely transcribed enhancers called in each cell line using our pipeline and then determined the level of transcription for each enhancer (Fig. 3A; Supplemental Fig. S2A) and the nearest neighboring gene (upstream or downstream) (Fig. 3B) or within 300 kb (Supplemental Fig. S2B). As expected, the uniquely transcribed enhancers exhibited high levels of transcription in the cell type in which they were active, but the same loci were minimally transcribed in all other cell lines (Fig. 3A; Supplemental Fig. S2A), which is an important control for this analysis. Likewise, the nearest neighboring gene for each uniquely transcribed enhancer exhibited high levels of transcription in the cell type in which the enhancers were active, but the same genes were minimally transcribed in all other cell lines (Fig. 3B; Supplemental Fig. S2B). These results suggest that the enhancers called using GRO-seq data in our pipeline are associated with cell-type–specific patterns of gene expression.

Actively transcribed enhancers dictate subtype-specific transcriptional programs. (A) Box plots of normalized GRO-seq read counts for enhancers uniquely transcribed in a single cell line compared to the transcription of the same genomic loci in all other cell lines. Asterisks indicate significant differences between the two conditions tested for each cell line (Wilcoxon rank-sum test, P < 0.05). Colored circles indicate the molecular subtype of each breast cancer cell line (for the color codes, see Fig. 2B). (B) Box plots of normalized GRO-seq read counts for the nearest neighboring genes to uniquely transcribed enhancers (from A) in a single cell line compared to the transcription of the same genes in all other cell lines. Asterisks indicate significant differences between the two conditions tested for each cell line (Wilcoxon rank-sum test, P < 0.05). (C) The nearest neighboring genes to the uniquely transcribed enhancers in each cell line are preferentially expressed in patient tumor samples of the same molecular subtype. (Left) ZR-75-1 cells represent the Luminal A breast cancer molecular subtype. The nearest neighboring genes to the uniquely transcribed enhancers for ZR-75-1 cells are more highly expressed in Luminal A patient tumor samples compared to the other tumors types. (Right) For comparison, MDA-MB-468 cells represent the TN basal breast cancer molecular subtype. The nearest neighboring genes to the uniquely transcribed enhancers in MDA-MB-468 cells are more highly expressed in TN basal tumor samples compared to the other tumor types. Observed differences are significant as determined by an ANOVA comparison of the means (P-value < 0.00001).
To further assess the broader biological significance, we determined the expression of these same genes in patient tumor samples. The genes nearest to the uniquely transcribed enhancers in the ZR-75-1 Luminal A cell line were preferentially expressed in the corresponding patient tumor samples of the same molecular subtype (Fig. 3C). Likewise, the genes nearest to the uniquely transcribed enhancers in the MDA-MB-468 TN basal cells were preferentially expressed in the corresponding patient tumor samples of the same molecular subtype (Fig. 3C). These results illustrate that enhancers called using our pipeline with GRO-seq data from Luminal A and TN breast cancer cell lines are associated with biologically relevant gene expression patterns in breast cancer patient samples.
A functional score of enhancer elements identifies subtype-specific enhancers and their cognate TFs
The analyses described above are a useful way to identify active enhancers and their putative target genes, but they tell us little about the TFs that drive enhancer formation or the relative activity of each enhancer. Thus, we sought to develop a method that would allow us to extract more detailed functional information from the genomic data. In this regard, we developed a computational pipeline that we call the Total Functional Score of Enhancer Elements (TFSEE), which integrates data from GRO-seq, RNA-seq, histone modification ChIP-seq (i.e., H3K27ac and H3K4me1), and motif searches, allowing for the simultaneous identification of active enhancers and their cognate transcription factors across all breast cancer cell lines (Fig. 4A). TFSEE integrates (1) the location and magnitude of enhancer activity based on enhancer transcription (GRO-seq); (2) the enrichment of enhancer-related histone modifications (ChIP-seq); (3) the level of TF expression (RNA-seq); and (4) the TF motif scores for each enhancer (Fig. 4A).
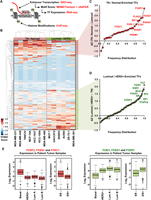
A functional score of enhancer elements identifies subtype-specific enhancers and their cognate TFs that drive subtype-specific gene expression in breast cancer cells. (A) Diagram of the data used for determining the Total Functional Score of Enhancer Elements (TFSEE) across breast cancer cell lines. TFSEE simultaneously identifies putative subtype-specific enhancers and their cognate TFs by integrating the magnitude of enhancer transcription (GRO-seq), TF mRNA expression levels (RNA-seq), TF motif P-values (MEME/Tomtom), and enrichment of H3K4me1 and H3K27ac (ChIP-seq). This analysis yields the location, activity level, and predicted TFs at each enhancer in all breast cancer cells. (B) Unsupervised hierarchical clustering of cell line–normalized TFSEE scores shown in a heatmap representation. Two major clades arise from this analysis, highlighting key TFs for TN/Normal subtypes versus Luminal/HER2+ subtypes. (C,D) Rank order frequency distribution of TFs enriched in the TN/Normal-like clade (C) and the Luminal/HER2+ clade (D) identified using TFSEE. The top TFs in each clade are noted. (E) Box plots of expression values for members of the Forkhead box family of TFs in patient breast tumor samples, confirming the differential enrichment of these TFs in the TN/Normal-Like versus the Luminal/HER2+ clades shown in C and D. Observed differences are significant as determined by an ANOVA comparison of the means (P-value < 0.00001).
For the de novo motif analyses, we searched a 1-kb region surrounding the transcribed enhancers for each cell line using MEME software (Bailey et al. 2009) and matched the motifs to known transcription factors from the Tomtom and JASPAR data bases (Supplemental Fig. S3; Gupta et al. 2007; Mathelier et al. 2016). We recorded the P-values for the motifs identified and assigned a higher score to more significant P-values. To ensure that the TFs whose motifs were called in our analysis were actually expressed in the corresponding cell lines, we used RNA-seq data to determine the expression levels of the mRNAs encoding them. Thus, the highest scoring TFs from TFSEE were those that had the (1) highest levels of enhancer transcription; (2) greatest enrichment of H3K4me1 and H3K27ac; (3) most significant P-values for the motifs; and (4) best correlation with the expression of the predicted TF in a given cell line.
We visualized the results from TFSEE using unsupervised hierarchical clustering (Fig. 4B), which grouped the cell lines into two major clades: (1) TN/normal; and (2) Luminal A+B/HER2+ (Fig. 4B). This analysis provided a clear demarcation of the TFs that were most enriched at transcribed enhancers between the two clades. Interestingly, previous gene expression profiling studies have also demonstrated an association or similarities between TN and “normal” cells, both of which do not express ER, PR, or HER2 and are technically triple negative (Charafe-Jauffret et al. 2006; Neve et al. 2006; Marcotte et al. 2016). We then determined a rank order frequency distribution for all TFs within each clade (Fig. 4C,D; Supplemental Table S1). One striking observation from this analysis was the abundance of Forkhead box family TFs that ranked high in both clades (Fig. 4C,D). Importantly, the clade-specific Forkhead TFs were selectively expressed in patient samples of the same molecular subtype (Fig. 4E). For example, FOXF2, FOXQ1, and FOXC1 were highly ranked in the TN/normal clade and are also more highly expressed in TN basal breast tumors compared to other molecular subtypes (Fig. 4E). The well-studied Forkhead TF FOXA1, which is expressed in luminal breast cancers (Bernardo et al. 2010; Hurtado et al. 2011), was highly enriched in the Luminal A+B/HER2+ clade, as expected, lending support to the fidelity of our approach. Similar results were obtained using pairwise comparisons of TFSEE scores from the different molecular subtypes, rather than unsupervised hierarchical clustering (Supplemental Figs. S4–S6). Collectively, the pairwise analyses identified most of the top TFs identified in the original unsupervised hierarchical clustering analysis (Supplemental Fig. S6A–D; cf. Fig. 4C,D).
To confirm experimentally that the predicted TFs did indeed bind to the transcribed enhancers, we performed ChIP-qPCR experiments for two transcription factors in each clade. These analyses revealed the enrichment of PLAG1 and RUNX2 (i.e., TN/normal clade) at transcribed enhancers in HCC-1937 TN cells (Fig. 5A; Supplemental Fig. S7A,B), as well as FOXA1 and HLF (i.e., Luminal A+B/HER2+ clade) at transcribed enhancers in MCF-7 Luminal A cells (Fig. 5B; Supplemental Fig. S7A,B). These TFs did not bind to a negative control region that exhibited no features indicative of an active enhancer (Fig. 5A,B; Supplemental Fig. S7A–D). Of the 24 potential positive or negative binding events tested for these four TFs (12 sites total tested in two cell lines), 22 (∼90%) gave the expected result. Taken together, our TFSEE analysis led to the identification of key enhancers and their cognate transcription factors that may control the gene expression programs that dictate the cellular phenotypes of the different molecular subtypes of breast cancers.
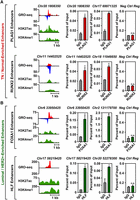
TFSEE-predicted transcription factors are enriched at sites of enhancer transcription. (A) Genome browser views of transcribed enhancers (GRO-seq; H3K27ac and H3K4me1) (left) and corresponding ChIP-qPCR experiments for their cognate predicted TFs (right) for two TFs highly enriched in the TN/Normal-Like clade based on TFSEE: PLAG1 (top) and RUNX2 (bottom). The data shown are from TN basal breast cancer cells (HCC-1937). Enhancer transcription provides a measure of activity and the locations of the enhancers, whereas ChIP-qPCR confirms the binding of the predicted TF at that site. The enhancers are designated by their genomic coordinates. A negative control region not bound by the predicted TFs is shown for comparison. Each bar represents the mean + SEM, n = 3. Asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (n.s.) not significant (Student's t-test, P-value > 0.05). (B) A set of experiments similar to those shown in A for two highly enriched TFs in the Luminal/HER2+ clade: FOXA1 (top) and HLF (bottom). The data shown are from Luminal A breast cancer cells (MCF-7). Each bar represents the mean + SEM, n = 3. Asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (n.s.) not significant (Student's t-test, P-value > 0.05).
FOS-like 1 (FOSL1) is enriched at transcribed enhancers in TN cells, regulates cellular proliferation, and is predictive of breast cancer patient outcomes
To confirm that subtype-specific TFs identified using TFSEE play a role in the biology of the cognate cell types, we performed a series of functional analyses on FOS-like 1 (FOSL1), a TF highly enriched in the TN/normal clade (Fig. 4C). FOSL1 is a member of the Fos gene family of leucine zipper proteins that dimerize with Jun proteins to form the AP-1 TF complex (Shaulian and Karin 2002; Shaulian 2010; Zhao et al. 2014). AP-1 is a key component of many signal transduction pathways, which regulate a variety of cellular processes including differentiation, apoptosis, migration, and transformation (Shaulian and Karin 2002; Shaulian 2010; Zhao et al. 2014).
To confirm that FOSL1 binds at predicted enhancers in TN cells (e.g., Fig. 6A; Supplemental Fig. S8A), we performed ChIP-qPCR at predicted enhancers in several TN cell lines. We observed a significant enrichment of FOSL1 at its cognate predicted enhancers, but not at a negative control region that exhibited no features indicative of an active enhancer (Fig. 6B; Supplemental Figs. S8B,C, S9). Of the 33 potential positive or negative FOSL1 binding events tested (11 sites total in three cell lines), 28 (∼85%) gave the expected result. Furthermore, siRNA-mediated knockdown of FOSL1 caused a significant reduction in enhancer transcription at the cognate predicted enhancers as measured by RT-qPCR (Fig. 6C). These results indicate that FOSL1 binds to the genomic loci and nucleates enhancer formation.
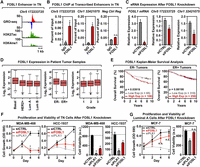
FOSL1 is enriched at transcribed enhancers in TN cells, regulates cell proliferation, and correlates with breast cancer patient outcomes. (A) Genome browser views of a transcribed enhancer predicted to be bound by FOSL1 in TN cells (GRO-seq; H3K27ac and H3K4me1). The data shown are from TN basal breast cancer cells (HCC-1937). (B) ChIP-qPCR for FOSL1 at two transcribed enhancers predicted to be bound by FOSL1, shown in a TN basal cell line (HCC-1937). The enhancers are designated by their genomic coordinates. Genome browser shots for the enhancer found on Chr 5 are shown in A. Each bar represents the mean + SEM, n = 3. Asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (C) siRNA-mediated knockdown of FOSL1 in a TN basal cell line (HCC-1937) decreases the transcription of cognate enhancers as determined by RT-qPCR. The enhancers are designated by their genomic coordinates. Each bar represents the mean + SEM, n = 3. Asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (D) Box plots of FOSL1 mRNA expression levels in patient tumor samples confirm enrichment of FOSL1 in Basal-like and in ER-negative (ER-) breast tumor samples, as predicted by the TFSEE analysis in breast cancer cell lines. Observed differences are significant as determined by an ANOVA comparison of the means (P-value < 0.00001). (E) FOSL1 mRNA expression is predictive of clinical outcomes in ER-negative (ER-) breast tumor patients. Kaplan-Meier survival analyses of patients expressing high levels of FOSL1 mRNA (maroon line) exhibit a poorer outcome compared to patients expressing low levels of FOSL1 mRNA (gray line). The breast cancer outcome-linked gene expression data were accessed and graphed using the Gene Expression-Based Outcome for Breast Cancer Online (GOBO) tool (Ringnér et al. 2011). (F) siRNA-mediated knockdown of FOSL1 in two TN cell lines (MDA-MB-468 and HCC-1937) causes decreased proliferation and viability, as observed in proliferation assays (left) and cell viability assays (right). siRNA-mediated knockdown of Polo-like Kinase 1 (PLK1) serves as a positive control. Each point or bar represents the mean + SEM, n = 3. Asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (G) siRNA-mediated knockdown of FOSL1 in Luminal A cell line (MCF-7) shows no significant effects on proliferation or viability compared to TN cells. siRNA-mediated knockdown of Polo-like Kinase 1 (PLK1) serves as a positive control. Each bar represents the mean + SEM, n = 3. The asterisks indicate significant differences from the corresponding control (Student's t-test, P-value < 0.05). (n.s.) not significant (Student's t-test, P-value > 0.05).
Extending these analyses to patient samples, we observed elevated expression of FOSL1 mRNA in TN basal tumors relative to other breast cancer subtypes, which increases with tumor grade (Fig. 6D). Furthermore, elevated expression of FOSL1 is associated with worse clinical outcomes (i.e., overall survival) in patients with ER-negative breast cancers, but not ER-positive breast cancers (Fig. 6E), suggesting a role for FOSL1 as an oncogene. Finally, to directly assess a subtype-specific functional role of FOSL1 in the biology of breast cancers, we knocked down FOSL1 in breast cancer cells and monitored the proliferation and viability of the cells, using knockdown of Polo-like Kinase 1 (PLK1) as a positive control. Knockdown of FOSL1 inhibited the proliferation and decreased the viability of several TN cell lines (Fig. 6F; Supplemental Fig. S8D,E), but had no effect on MCF-7 Luminal A cells (Fig. 6G).
A parallel set of experiments with PLAG1, another TF highly enriched in the TN/normal clade (Fig. 4C), confirmed that PLAG1 also plays a key role in enhancer formation and the biology of TN cells (Supplemental Fig. S10). In addition, we found that elevated expression of three other TFs in the TN/normal clade (i.e., PRDM1, IRF1, and RUNX2) is associated with better clinical outcomes (i.e., distant metastasis-free survival) in patients with ER-negative breast cancers, but not ER-positive breast cancers (Supplemental Fig. S11), suggesting roles for these TFs as tumor suppressors. Taken together, our results show that TFSEE can be used to identify breast cancer subtype-specific TFs that control the biology of those subtypes. In addition, our analyses led to the discovery of TN-specific TFs, such as FOSL1, that control the proliferation and viability of TN cells and whose expression is predictive of clinical outcomes in patients.
Discussion
The results from our analyses demonstrate that enhancer transcription can be used to improve the fidelity of functional enhancer identification. In addition, our study identified TFs that play critical roles in the biology of specific types of breast cancers, including Forkhead TFs, FOSL1, and PLAG1. More broadly, our study described a genomic and computational approach for identifying enhancers and their cognate TFs that play a critical role in the biology of particular cell types, which should be applicable to a wide variety of biological systems.
Enhancer transcription defines functional regulatory elements and their cognate TFs
We and others have shown previously that enhancer transcription, as defined by GRO-seq and related methods (e.g., PRO-seq), is a robust way to identify regulatory elements, such as enhancers, on a global scale (Hah et al. 2011, 2013; Wang et al. 2011; Core et al. 2014; Chae et al. 2015; Heinz et al. 2015). In addition, we have shown that the presence of enhancer transcription can be used to distinguish between active enhancers and inactive TF binding sites (Hah et al. 2013; Franco et al. 2015). Other genomic methods that have been used to identify enhancers include (1) DNase-seq, which measures accessibility (hypersensitivity) of the genome (although indiscriminately between open promoters, gene bodies, and enhancers); (2) ChIP-seq for histone modifications typically enriched at enhancers, such as H3K4me1 and H3K27ac; and (3) ChIP-seq for common enhancer-enriched coregulators, such as the acetyltransferases EP300 (a.k.a., p300) and CBP (Shlyueva et al. 2014). A directed approach using ChIP-seq for a TF of interest can also be used, but this approach is biased and requires prior knowledge about the TFs that are functioning in a given cell type. These approaches yield thousands of putative enhancers (e.g., Fig. 2E); the challenge is determining which of the putative enhancers are functional in the cell type of interest.
TFSEE using GRO-seq data allows for the simultaneous identification of enhancers, assessment of enhancer activity and gene expression, and identification of TFs that drive cell-type–specific gene expression programs (Fig. 4A). TFSEE improves enhancer identification by focusing the analysis on those enhancers that are most likely to be functionally active in a given cell type. Although TFSEE calls fewer enhancers from GRO-seq data than is typically called using other types of enhancer-related genomic data (Fig. 2E), the enhancers called from GRO-seq data exhibit the greatest overlap with enhancers called using other approaches (Fig. 2D). Transcribed enhancers called by three or four genomic assays are more likely to be functional in the cell type in which they are called than enhancers called by a single genomic assay. Moreover, the functional indications from enhancer transcription, which are not evident with other enhancer features (e.g., DNase I hypersensitivity, histone modification enrichment), increase the likelihood of identifying functional enhancers, as opposed to inactive TF binding sites.
Forkhead family TFs in luminal and triple negative breast cancers
Our analyses identified TFs that play subtype-specific roles in breast cancers, including Forkhead TFs. The Forkhead (FOX) proteins comprise a diverse family of TFs with roles in a variety of biological systems, including cancers (Benayoun et al. 2011; Lam et al. 2013). We found FOXF2, FOXQ1, and FOXC1 to be enriched in TN and ER-negative breast tumors compared to other molecular subtypes, whereas FOXI1, FOXA1, and FOXP2 were enriched in luminal and ER-positive breast tumors compared to other molecular subtypes (Fig. 4E). The FOX-related results from our TFSEE analyses are well supported by the literature. For example, previous studies have shown that FOXC1 regulates basal-like breast cancer cells by activating NF-κB signaling (Wang et al. 2012) and predicts poor overall survival in this breast cancer subtype (Ray et al. 2010). Likewise, FOXA1 is a well-characterized TF critical for the estrogen-dependent growth and proliferation of luminal breast tumors (Bernardo et al. 2010; Hurtado et al. 2011). Our results suggest that additional exploration of the functions of the Forkhead family of TFs in breast cancers is warranted.
FOSL1 and PLAG1: TFs driving the triple negative breast cancer phenotype
Our studies also identified and verified FOSL1 (a.k.a., fra-1) as a key TF in the biology of TN cells. In this regard, we found that FOSL1 enhancers are enriched in TN cells (Fig. 4C), and knockdown of FOSL1 inhibits the proliferation and viability of TN cells (Fig. 6F). Furthermore, FOSL1 expression is elevated TN basal tumors (Fig. 6D), a feature that is associated with poor clinical outcomes in patients (Fig. 6E). Thus, FOSL1 exhibits hallmarks of a cancer “driver,” which may be particularly active in TN cells. FOSL1 dimerizes with Jun proteins to form AP-1 TF complexes (Shaulian and Karin 2002; Shaulian 2010; Zhao et al. 2014), which function as key regulators of gene expression in cancers (Eferl and Wagner 2003; Verde et al. 2007). FOSL1 has previously been implicated in a variety of cancer types, including those of the colon, lung, and breast (Young and Colburn 2006). FOSL1 acts to induce epithelial-to-mesenchymal transitions and drive metastasis in breast and other cancers (Diesch et al. 2014; Risolino et al. 2014; Bakiri et al. 2015; Dhillon and Tulchinsky 2015; Iskit et al. 2015; Liu et al. 2015).
Finally, our studies identified and verified PLAG1 as a key TF in the biology of TN cells (Supplemental Fig. S10). PLAG1 (Pleomorphic adenoma gene 1 protein) is a zinc-finger transcription factor, which has been implicated in cancer (Abdollahi 2007; Van Dyck et al. 2007). Unlike FOSL1, little if any previous direct evidence exists linking PLAG1 to breast cancers. Rather, the gene encoding PLAG1 is consistently rearranged and subject to activating reciprocal chromosomal translocations involving 8q12 in pleomorphic adenomas of the salivary glands (Kas et al. 1997; Abdollahi 2007; Van Dyck et al. 2007). Thus, TFSEE can be a useful approach for identifying new TF drivers in cancers.
Collectively, our results provide a clear example of how integrative computational analyses of genomic data can be used to understand the molecular mechanisms supporting the function and biology of specific cell types.
Methods
Cell culture
All cell lines were purchased from the American Type Culture Collection (ATCC) and were maintained, propagated, and plated for experiments in the laboratory of Dr. Khandan Keyomarsi at the MD Anderson Cancer Center. Cell proliferation was assessed using a crystal violet staining assay, and cell viability was assessed using CellTiter-Glo reagent (Promega) for cells transfected with siRNAs using reverse transfection methodology. Additional details about the cell culture conditions, cell proliferation and viability assays, and siRNA-mediated knockdown are provided in the Supplemental Methods.
Kaplan-Meier and gene expression analyses in patient tumor samples
Kaplan-Meier estimators (Kaplan and Meier 1958; Dinse and Lagakos 1982) were generated using the Gene Expression-Based Outcome for Breast Cancer Online (GOBO) tool (http://co.bmc.lu.se/gobo/) (Ringnér et al. 2011) and the KM Plotter Tool (Szasz et al. 2016). Gene expression levels in patient tumor samples were also obtained using the GOBO tool.
Global run-on sequencing (GRO-seq)
Cells were collected at ∼70%–80% confluence, and nuclei were isolated as described previously (Luo et al. 2014). Nuclear run-on and GRO-seq library preparation were performed as previously described (Hah et al. 2011), with modifications (Danko et al. 2013; Luo et al. 2014). After library quality control assessment using a Bioanalyzer (Agilent), the samples were subjected to 50-bp single-end sequencing using an Illumina HiSeq 2000 Sequencing System. Additional details about the preparation of nuclei, nuclear run-ons, GRO-seq library preparation, and sequencing are provided in the Supplemental Methods.
Analysis of GRO-seq data
The GRO-seq data were analyzed using the groHMM package as described previously (http://bioconductor.org/packages/release/bioc/html/groHMM.html) (Hah et al. 2011; Luo et al. 2014; Chae et al. 2015) and the approaches described below. Quality control for the GRO-seq data was performed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). GRO-seq reads were trimmed to remove adapter contamination and poly(A) tails using the default parameters of Cutadapt software (Martin 2011). Reads >32 bp long were retained for alignment to the human reference genome using the BWA aligner v 0.6.1 (Li and Durbin 2010). Transcript calling was performed using groHMM, a two-state hidden Markov model–based algorithm as described previously (http://bioconductor.org/packages/release/bioc/html/groHMM.html) (Hah et al. 2011; Chae et al. 2015) on each individual cell line. Enhancer transcripts were classified into (1) short paired eRNAs; and (2) short unpaired eRNAs, as described previously (Hah et al. 2013). The universe of expressed eRNAs (short paired and short unpaired) was assembled and used for further analyses.
The universe of expressed genes in each cell line was determined from GRO-seq data using an RPKM cutoff ≥2. The set of nearest neighboring expressed genes for each enhancer defined by an expressed eRNA was determined for each cell line. De novo motif analyses were performed on a 1-kb region (±500 bp) surrounding the peak summit or the transcription start site for short paired and short unpaired eRNAs, respectively, using MEME (Bailey et al. 2009). The predicted motifs were matched to known motifs using Tomtom (Gupta et al. 2007).
Additional details about quality control and trimming, read alignment and gene annotation, transcript calling using groHMM, enhancer transcript calling, nearest neighboring gene analyses, motif analyses, and the generation of box plots are provided in the Supplemental Methods.
RNA isolation, RT-qPCR, and RNA-seq
Cells were collected at ∼70%–80% confluence, and total RNA for RT-qPCR and RNA-seq was performed using the RNeasy Mini Kit (Qiagen). Changes in the expression of eRNAs and mRNAs were analyzed by RT-qPCR, as previously described (Franco et al. 2015). mRNA-seq libraries were prepared using methods described previously (Zhong et al. 2011). After library quality control assessment using a Bioanalyzer (Agilent), the samples were subjected to 50-bp single-end sequencing using an Illumina HiSeq 2000 Sequencing System. At least two biological replicates were sequenced for each cell line to achieve a minimum of roughly 65 million raw reads per cell line. Additional details about RNA isolation, RT-qPCR and primers, RNA-seq library preparation, and sequencing are provided in the Supplemental Methods.
Analysis of RNA-seq data
The raw data were subjected to QC analyses using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were then mapped to the human reference genome using the default parameters in TopHat (v2.0.12) (Trapnell et al. 2009). For expression analyses, FPKM values were calculated per gene using Cufflinks (v.2.1.2) (Trapnell et al. 2010). Additional details about quality control and trimming, read alignment, and expression analyses are provided in the Supplemental Methods.
ChIP-qPCR and ChIP-seq
ChIP was performed as previously described (Franco et al. 2015) with a few modifications. Cells were grown to ∼70%–80% confluence and then cross-linked with 1% formaldehyde for 10 min at 37°C. ChIPed DNA was analyzed by qPCR using enhancer- or gene-specific primers, or used for ChIP-seq. ChIP libraries were prepared using a modified Kapa LTP Library Preparation kit (KAPA Biosystems) for Illumina Platforms (Y Xi, W Li, K Tanaka, KL Allton, D Richardson, J Li, HL Franco, A Nagari, V Malladi, LD Coletta, et al., in prep.). The quality of the final libraries was assessed using a 2200 TapeStation (Agilent Technologies). The libraries were sequenced using a HiSeq 2500 sequencer (Illumina; Single-end reads, 36 bp for all samples). At least two biological replicates were sequenced for each cell line for a minimum of roughly 100 million raw reads per cell line. Additional details about the antibodies used for ChIP, the ChIP method, qPCR and primers, ChIP-seq library preparation, and sequencing are provided in the Supplemental Methods.
Analysis of ChIP-seq data
The raw reads were aligned to the human reference genome using default parameters in Bowtie (ver. 1.0.0) (Langmead et al. 2009). The aligned reads were subsequently filtered for quality and uniquely mappable reads using SAMtools (version 0.1.19) (Li et al. 2009) and Picard (version 1.127; http://broadinstitute.github.io/picard/). Relaxed peaks were called using MACS (v2.1.0) (Feng et al. 2012) with a P-value = 1 × 10−2. Additional details about quality control and trimming, read alignment, and peak calling are provided in the Supplemental Methods.
Predicting breast cancer subtype-specific TFs using TFSEE
We developed a pipeline in Python (Total Functional Score of Enhancer Elements [TFSEE]) that combines GRO-seq, RNA-seq, and ChIP-seq data with TF motif information to predict TF driving the formation of active enhancers in each breast cancer cell line, as well as the locations of the cognate enhancers (the scripts are included in a Supplemental File). The algorithm does the following (for details, see Supplemental Material): (1) normalizes enhancer expression using GRO-seq; (2) normalizes enhancer expression using ChIP-seq; (3) determines enhancer activity; (4) normalizes motif predictions; (5) normalizes TF expression using RNA-seq; and (6) determines the Total Functional Score of Enhancer Elements (TFSEE) and generates a heatmap.
Additional details about (1) normalization of enhancer activity, motif predictions, and transcription factor expression; (2) defining total enhancer activity; (3) determining the total functional score of enhancer elements (TFSEE); (4) pairwise Pearson's correlation analyses; and (5) the generation of heatmaps are provided in the Supplemental Methods.
Oligonucleotide sequences
The sequences of all oligonucleotides used for RT-qPCR, ChIP-qPCR, and siRNA-mediated knockdown are provided in the Supplemental Methods.
Data access
Deep sequencing data from this study are available from the NCBI's Gene Expression Omnibus repository (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the following accession numbers: GSE96859 (GRO-seq), GSE96860 (RNA-seq), and GSE85158 (ChIP-seq). The custom scripts for TFSEE are provided in the Supplemental Material.
Acknowledgments
We thank Xiaole Shirley Liu, Jean-Pierre Issa, Brad Cairns, Jeff Rosen, and members of the Kraus laboratory, for helpful comments and discussions. This work was supported by a grant from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP110471-P1) to S.Y.R.D., W.L.K., Wq.L., X.S., M.T.B., M.C.B., and K.K.; a grant from the NIH/National Cancer Institute (R00 CA204628) to H.L.F.; and a grant from NIH/National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK058110) to W.L.K.
Author contributions: PIs of the LONESTAR Consortium Project 1 (S.Y.R.D., W.L.K., W.L., X.S., M.T.B., M.C.B., K.K.) conceived of the overall project and directed its execution in their laboratories. H.L.F., A.N., V.M., and W.L.K. developed the specific project described herein with input from S.Y.R.D., W.L., X.S., M.T.B., M.C.B., and K.K. and performed the experiments and data analyses in the Kraus laboratory. H.L.F. generated the GRO-seq and RNA-seq data sets and performed the cellular and molecular assays. V.M., A.N., and H.L.F. developed TFSEE with input from W.L.K.; processed and analyzed the GRO-seq and RNA-seq data; and performed the integrative computational analyses of the genomic data. D.R. and K.K. grew the cells and ensured quality control. Wq.L., K.L.A., J.L., K.T., S.M., M.T.B., X.S., M.C.B., and S.Y.R.D. generated the ChIP-seq data sets. Y.X. and Wq.L. processed the ChIP-seq data and performed the initial analyses. All authors contributed to the data interpretation and presentation. H.L.F., V.M., and A.N. prepared the figures and wrote the methods, and H.L.F. wrote the remaining text. The figures and text were edited and finalized by W.L.K. with input from H.L.F., V.M., A.N., and the rest of the group. A description of the LONESTAR Consortium is provided in the Supplemental Materials.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.226019.117.
- Received June 6, 2017.
- Accepted December 19, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








