
SelexGLM differentiates androgen and glucocorticoid receptor DNA-binding preference over an extended binding site
- Liyang Zhang1,5,
- Gabriella D. Martini2,3,5,
- H. Tomas Rube2,3,
- Judith F. Kribelbauer2,3,
- Chaitanya Rastogi2,3,
- Vincent D. FitzPatrick2,3,
- Jon C. Houtman4,
- Harmen J. Bussemaker2,3 and
- Miles A. Pufall1
- 1Department of Biochemistry, Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242, USA;
- 2Department of Biological Sciences, Columbia University, New York, New York 10027, USA;
- 3Department of Systems Biology, Columbia University Medical Center, New York, New York 10032, USA;
- 4Department of Immunology, Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242, USA
-
↵5 These authors contributed equally to this work.
Abstract
The DNA-binding interfaces of the androgen (AR) and glucocorticoid (GR) receptors are virtually identical, yet these transcription factors share only about a third of their genomic binding sites and regulate similarly distinct sets of target genes. To address this paradox, we determined the intrinsic specificities of the AR and GR DNA-binding domains using a refined version of SELEX-seq. We developed an algorithm, SelexGLM, that quantifies binding specificity over a large (31-bp) binding site by iteratively fitting a feature-based generalized linear model to SELEX probe counts. This analysis revealed that the DNA-binding preferences of AR and GR homodimers differ significantly, both within and outside the 15-bp core binding site. The relative preference between the two factors can be tuned over a wide range by changing the DNA sequence, with AR more sensitive to sequence changes than GR. The specificity of AR extends to the regions flanking the core 15-bp site, where isothermal calorimetry measurements reveal that affinity is augmented by enthalpy-driven readout of poly(A) sequences associated with narrowed minor groove width. We conclude that the increased specificity of AR is correlated with more enthalpy-driven binding than GR. The binding models help explain differences in AR and GR genomic binding and provide a biophysical rationale for how promiscuous binding by GR allows functional substitution for AR in some castration-resistant prostate cancers.
Gene expression programs are precisely regulated by transcription factors (TFs), a class of DNA-binding proteins that orchestrate the activity of the RNA polymerase II and chromatin-modifying complexes. The DNA-binding domains (DBDs) of TFs fall into families consisting of dozens or even hundreds of members (Weirauch et al. 2014), leading to similar DNA sequence preferences among family members. Nonetheless, subtle quantitative differences in DNA-binding specificity between related TFs are associated with large qualitative differences in the sets of target genes they control (Maerkl and Quake 2007). To understand gene regulation and regulatory networks, it is therefore essential not only to accurately quantify these differences in DNA recognition but also to determine the structural and physical basis of that specificity. The former can be done using comprehensive, unbiased experimental and computational methods; the latter requires more focused mechanistic analyses.
The intrinsic DNA-binding specificities for hundreds of TFs have been profiled using a number of different high-throughput assays. These include (universal) protein binding microarrays (PBMs) (Berger and Bulyk 2009), bacterial one-hybrid (B1H) (Meng et al. 2005), and (solution-based) high-throughput SELEX (HT-SELEX) (Zhao et al. 2009; Jolma et al. 2010, 2013). These methods have generated rich data sets that have been exploited to define DNA-binding ‘motifs’ for all major TF families and capture differences in DNA sequence preference within these families. DNA-binding specificities can be represented in the form of a position weight matrix (Stormo 2000) or a position-specific affinity matrix (PSAM) (Foat et al. 2006), which in turn can be visualized as an information content logo (Schneider et al. 1985) or energy/affinity logo (Foat et al. 2006). The motifs are available through databases such as JASPAR (Mathelier et al. 2015), UniPROBE (Orenstein and Shamir 2014; Hume et al. 2015), and Cis-BP (Weirauch et al. 2014).
Sequences outside the core motif can also contribute to DNA-binding specificity. In particular, flanking A and T homopolymers can cause increased affinity (Jolma et al. 2013; Levo et al. 2015). This can be due to a narrowing of the minor groove, which attracts positively charged basic residues (Rohs et al. 2009b), but in other cases, the mechanism is unclear (Dror et al. 2015). Flanking sequences are also increasingly being recognized as a vehicle for diversified binding preference among paralogous TFs (Fisher and Goding 1992; Maerkl and Quake 2007; Zhou and O'Shea 2011; Slattery et al. 2014). For example, Cbf1p and Tye7p, members of the basic helix-loop-helix (bHLH) family in Saccharomyces cerevisiae, recognize similar E-box core sequences (CANNTG), but distinct flanking preferences became apparent when binding was assayed over a larger footprint using custom-designed genomic context PBMs (gcPBMs) (Gordân et al. 2013). Similar effects of flanking sequences on specificity have been observed among ETS proteins using PBMs (Wei et al. 2010). Moreover, SELEX-seq technology has been used to show that Hox proteins read out the spacer sequences between half-sites in distinct ways when binding as heterodimers with the cofactor Exd (Slattery et al. 2011; Abe et al. 2015).
The importance of being able to understand functional differences between close TF paralogs is brought into sharp relief by castration-resistant prostate cancer (CRPC). Prostate cancer is driven by androgen signaling through regulation of gene expression by the androgen receptor (AR; AR) (Watson et al. 2015). Blocking of androgen synthesis and inhibition of ligand binding to AR have both been effective treatments. Unfortunately, CRPC eventually arises due to alternative production of androgens or activation of AR (Feldman and Feldman 2001). In some cases, CRPC is accompanied by increased expression of the glucocorticoid receptor (GR; NR3C1), which then functionally substitutes for AR by activating a subset of the AR transcriptional program that drives cancer progression (Arora et al. 2013). Despite this overlap, chromatin immunoprecipitation followed by sequencing (ChIP-seq) in LNCaP-1F5, a cell line model of CRPC, has shown that AR and GR share only about a third of their genomic binding sites (Sahu et al. 2013, 2014). Although cofactors such as the TF FoxA1 help distinguish between AR and GR binding (Sahu et al. 2013; Belikov et al. 2016), these two factors still bind distinct loci in their absence, suggesting an intrinsic ability to distinguish sequences that are not reflected in previous measurements of their in vitro specificities (He et al. 2012; Jolma et al. 2013; Jin et al. 2014; Pihlajamaa et al. 2014).
It is not clear how AR and GR, both members of the steroid hormone receptor (SHR) family, are directed to different genomic loci. Existing ChIP-seq and HT-SELEX (Nelson et al. 1999; Jolma et al. 2013; Sahu et al. 2013; Yang et al. 2017) have been unable to detect consistent differences in how AR and GR distinguish sequences within or outside a 15-bp core motif composed of inverted hexameric half-sites separated by a 3-bp spacer: RGAACANN NTGTTCY. Crystallographic evidence indicates that AR and GR each bind DNA as head-to-head dimers, with the two monomer subunits each occupying a half-site and dimerizing over the spacer (Shaffer et al. 2004; Meijsing et al. 2009; Watson et al. 2013). Our detailed analysis of AR- and GR-DNA crystal structures indicates that they present identical amino acids at the DNA-binding interface (Supplemental Fig. S1). Conserved residues make specific contacts in the major groove at positions 2, 4, and 5 (Supplemental Fig. S2; Luisi et al. 1991; Arbuckle and Luisi 1995), accounting for most of the binding energy, although other noncontacted base pairs within the half-site have also been shown to affect GR affinity (La Baer and Yamamoto 1994). Additional energy is derived from backbone contacts along the 3-bp spacer, which are sensitive to minor groove width in GR (Meijsing et al. 2009; Watson et al. 2013). Contacts made with DNA sequences flanking the core motif further contribute to GR affinity (Meijsing et al. 2009). In this work, we test whether, despite their conservation, AR and GR use this shared DNA-binding interface differently to distinguish sequences within and flanking the core 15-bp motif.
Motif discovery in DNA sequences has a long history that includes the Gibbs sampler (Thompson 2003), the MEME algorithm (Bailey and Elkan 1994; Ma et al. 2014), and the application of a profile/hidden Markov model (HMM)–based method to SELEX data (Roulet et al. 2002). These traditional ‘probabilistic’ algorithms were designed to detect and characterize base preference patterns in sets of unaligned DNA sequences. There is in principle no limit to the size of the motifs that can be discovered. Unfortunately, the algorithms in this class were not designed, and are not suitable, for building accurate quantitative models from high-throughput functional genomics data (Bussemaker et al. 2007; Ruan and Stormo 2017). For instance, the penalty associated with base substitutions away from the optimal sequence in the models these algorithms produce strongly depends on the criterion that was used to define the set of ‘bound’ sequences. This is a fundamental problem, as DNA-binding specificity is a quantitative property of the TF protein, whose estimate should not depend on how a training set of DNA sequences was defined. Furthermore, although the log-likelihood scores produced by probabilistic motif discovery algorithms are often used as a surrogate for true binding free-energy differences (Berg and Hippel 1987), they are at best defined up to an overall scaling factor. This is also problematic, because it leaves the fold-change in affinity that is associated with base substitutions ill defined. Finally, a recent algorithm based on deep learning (Alipanahi et al. 2015) performed well on a classification task but was not trained in a way that was designed to make quantitative predictions.
In view of the above concerns, it is desirable that an algorithm for inferring an accurate binding model from HT-SELEX data has its foundation in a biophysical description of the protein–DNA interaction in the context of the SELEX assay (Djordjevic et al. 2003; Zhao et al. 2009; Djordjevic 2010; Atherton et al. 2012; Ruan and Stormo 2017). In practice, however, estimating binding model coefficients from SELEX data remains a challenging numerical problem. To make the binding model inference problem tractable, existing biophysically inspired algorithms use approximations and rely on prior sequence-based alignment of the sequences (Djordjevic and Sengupta 2006; Stormo and Zhao 2010). Some other approaches to SELEX analysis start by tabulating the relative enrichment between rounds for all oligomers of a given length (Jolma et al. 2010, 2013; Slattery et al. 2011; Riley et al. 2014). These methods also yield imperfect estimates of relative binding affinities because they do not explicitly consider where the TF prefers to bind within each DNA probe.
The recently published BEESEM algorithm (Ruan et al. 2017) addresses binding position preference within the probe explicitly within the context of a biophysical model of SELEX. However, the numerical procedure BEESEM uses to estimate binding model parameters is based on minimization of the least-squares error between observed and expected values across all of a round-to-round enrichment metric that weighs k-mer occurrences according to the relative probability with which the TF binds each position with a particular probe (Ruan et al. 2017). As such, BEESEM still relies on enumeration of all k-mers, which in practice limits its application to a motif width of 12 bp. Thus, to the best of our knowledge, no published algorithm currently exists that is both (1) based on a biophysical (as opposed to a probabilistic) approach and therefore has the potential of yielding accurate estimates of binding free energies and (2) capable of modeling binding specificity over a large enough footprint to accommodate the flanks of the AR or GR binding site outside the 15-bp core.
The SelexGLM algorithm that we introduce in this work and use to analyze the SELEX-seq data for AR and GR builds on the existing idea of using a biophysical model to identify the preferred position where the protein binds within each probe (Atherton et al. 2012; Ruan and Stormo 2017). However, SelexGLM differs from these in that it ignores probes whose selection is not dominated by a single binding site within them. In addition, SelexGLM uses a generalized model based on the Poisson distribution to estimate the binding free-energy coefficients, which allows us to perform fits even at very low probe counts. These features lift the practical restriction on footprint size that other biophysically motivated algorithms are (implicitly) limited by. Here we show how this enables us to uncover and carefully characterize intrinsic differences in DNA-binding specificity between AR and GR.
Results
AR interacts with DNA over a larger footprint than GR
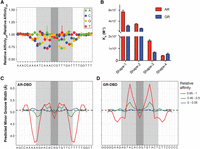
To determine the intrinsic specificity of AR and GR at high resolution, we performed SELEX-seq (Slattery et al. 2011; Riley et al. 2014) for homodimers of the DBD of each factor (Fig. 1A; Supplemental Fig. S1C). Although SELEX-seq (and lower throughput predecessors) have been developed over the years (Djordjevic 2010; Ogawa and Biggin 2011), increased sequencing and computational power have allowed some refinements. Purified proteins were incubated with a pool of DNA molecules, each containing a larger (23-bp) random region than typical, flanked by Illumina adapters and tagged at one end with Cy5. Electrophoretic mobility shift assays (EMSAs) were used to separate dimer-bound DNA sequences over eight rounds of affinity-based selection (Fig. 1A; Supplemental Fig. S3). After each round, the concentration of the isolated DNA was quantified by qPCR and then in parallel amplified for reselection and packaged into a sequencing library by adding Illumina flow cell adapters. Each library was then sequenced to a depth of about 107 reads. Preliminary analysis of the SELEX-seq data revealed unexpected differences between AR and GR. Following a previous study (Slattery et al. 2011), we estimated affinities as normalized oligomer enrichments, using the R/Bioconductor package SELEX (Riley et al. 2014). Biases in the initial round zero (R0) pool were estimated using a fifth-order Markov model, after which we computed the information gain between the initial (R0) and final round (R8) to estimate binding site size. For GR, the information gain peaks at 15 bp (Fig. 1C), consistent with the previously defined core motif. However, for AR, information content continues to increase beyond 15 bp (Fig. 1D), indicating a sensitivity to base identity over a larger binding site. One concern was that sequences would be overselected after eight rounds. Due to the diversity of the pool and lack of a strict consensus sequence for both AR and GR, very few 23-mers sequences are observed more than once in the sequenced libraries (Fig. 1E). This indicates not only that the libraries were not overselected but also that further rounds of selection might allow better discrimination of lower affinity sequences without overselecting high-affinity sites (Djordjevic 2010). A cursory k-mer–based analysis indicated that AR and GR differ in their specificities, sharing only about a third of their significantly enriched 15-mers (Fig. 1F; Supplemental Fig. S4A,B), with substantially different preferences among those common 15-mers (Fig. 1G) and confirmed specificity for AR over a larger footprint, of at least 21 bp (Supplemental Fig. S4C), than GR (Supplemental Fig. S4D). Quantitative EMSAs supported the rank order of these sequences in terms of their enrichment in the SELEX-seq experiment (Supplemental Fig. S4E,F).
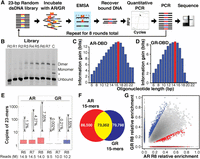
SELEX-seq reveals differences in AR- and GR-DBD (DNA-binding domain) DNA-binding specificity. (A) SELEX-seq. A 70-bp dsDNA library with 23-bp randomized region was incubated with the DBD of AR or GR and separated into monomer and dimer species by EMSA. Dimer-bound DNA was recovered, quantified by qPCR, amplified as the library for the next round, and repeated for eight rounds. Each round of library, including the initial dsDNA library, was sequenced. (B) EMSA gel showing the enrichment of dimer-bound sequences after each round of selection for GR-DBD. The intensity of the shifted band plateaus after round 7. A high-affinity palindromic sequence served as a control to locate the dimer band. (*) An artifact during the synthesis of control sequence but not observed in the SELEX library. (C,D) Information gain, or Kullback-Leibler divergence, from R0 to R8, as a function of oligonucleotide length. (E) Boxplot showing the multiplicity of unique 23-mers in each of the last three rounds of SELEX-seq selection for AR and GR. Even for the most highly selected library (AR R8) fewer than 10% of all reads have 10 copies or more, indicating that the libraries are not overselected. (F) Venn diagram showing the overlap of sequences for AR- and GR-DBD with at least 100 sequencing counts. (G) Scatterplot of sequences that were commonly bound (yellow from F) by AR- and GR-DBD.
Quantifying DNA-binding specificity using feature-based modeling
To overcome the limitations of existing algorithms that prevented us from inferring an accurate binding model from our SELEX-seq
data for AR and GR over their entire (>20 bp) footprint, we developed SelexGLM, a flexible modeling strategy based on Poisson regression that allows us to estimate binding free-energy contributions throughout
the protein–DNA interface directly from the SELEX-seq read counts (Fig. 2). We use a standard equilibrium thermodynamics description of protein–DNA interaction that was previously used to analyze
PBM data (Foat et al. 2006; Zhao and Stormo 2011; Gordân et al. 2013; Riley et al. 2015). The relative binding free energy for sequence S is modeled as a sum of parameters ΔΔGϕ associated with the DNA sequence features ϕ ∈ Φ(S) that characterize the difference between S and a reference sequence Sref (typically the highest-affinity sequence):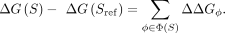
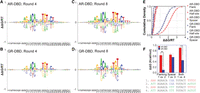
SelexGLM shows differences in DNA recognition between AR and GR throughout their binding sites. (A–D) Energy logos for AR-DBD (top) and GR-DBD (bottom), obtained by fitting biophysical models for protein–DNA interaction to the SELEX read counts using an iterative generalized linear modeling approach based on Poisson regression, implemented as SelexGLM. Highly similar logos were obtained using two separate rounds of data. See Supplemental Figure S5 for logos generated using round 4 to 8 data. (E) Cumulative distribution functions for the contribution of half-site (squares), spacer (triangles), and flanking (circles) sequences on AR-DBD (red) and GR-DBD (blue) binding energy. (F) Validation of the contribution of flanking A tracts and spacer to AR- and GR-DBD binding performed by quantitative electrophoretic mobility shift assay (EMSA). Loss of flanking A tracts is more detrimental to AR- than GR-DBD (one vs. two), whereas changing spacer can have detrimental effects on the binding of both (one vs. three). Error bars, SEM based on at least three repeats of each experiment. (*) P-value ≤0.05, (**) P-value ≤0.01, two-sided t-test.
In this study, we restricted ourselves to single-nucleotide features (e.g., ϕ = C3, denoting the presence of a C at position 3 within the binding site window). Our modeling assumptions imply that the combined effect of multiple mutations within the binding site is additive in terms of binding free energy or multiplicative in terms of relative affinity.
The model parameters are found through an iterative fitting process that starts from an initial estimate, or seed. To generate this seed, we construct a relative enrichment table using a footprint large enough to capture the binding site core but not so large that counts get too low. We chose a seed length of 15 bp for both AR and GR, but the final model is only weakly dependent on this choice and can have a much larger footprint (31 bp in our case; see below). The negative logarithm of the relative enrichment of each mutated 15-mers is used as an initial estimate of ΔΔGϕ/RT for each feature.
Once seeded, the model is refined by alternating between two steps. In the first step, we determine the highest-affinity binding
site within each unique observed SELEX probe in the data (“affinity-based alignment”). This allows us to construct a design
matrix X defining each DNA feature (in this case each base pair) relative to the optimal binding window in each probe; only probes
whose rate of selection is dominated by a single binding site offset are included (see Methods). In the second step, the design
matrix is used to fit a generalized linear model (GLM) to the read counts, leading to a re-estimated set of free-energy coefficients: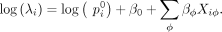
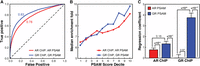
Here λi is the expected value of the read count y for probe i: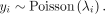
Each model coefficient βϕ is used as a re-estimate of ΔΔGϕ/RT, which are then used to update the design matrix, and the process repeats. Convergence is reached once the position of the optimal binding window no longer changes for any of the probes in the data set after re-estimation of the free-energy coefficients.
Earlier rounds of selection are sufficient for SelexGLM analysis
We originally performed SELEX-seq over eight rounds of selection in order to obtain linear enrichment of sequences appropriate for oligomer enrichment based analysis. The most enriched 21-mer for AR (AAAAGAACACGATGTACTTTT) is contained in approximately 4000 reads out of the ∼107 that make up the R8 library. Most suboptimal sequences of the same length will not be present in the library as their expected count decreases exponentially with the number of rounds. This, however, is not a problem for SelexGLM, which uses Poisson regression techniques to deal with low read counts. When we analyzed each round of selection separately using SelexGLM, we found that, although R8 provided the highest-resolution model without over selection (Fig. 1E; Supplemental Fig. S5A–J), high-quality models could also be generated from earlier rounds of selection (Supplemental Fig. S5A–J). However, the accuracy of the models increased in the later rounds, particularly when it comes to distinguishing the base-pair substitutions with the most deleterious effect on binding (Supplemental Figs. S5, S6). Thus, we used models generated from R8 for both proteins in all subsequent analyses.
AR is distinguished from GR by a preference for poly(A) sequences outside the 15-bp core
We used SelexGLM to build recognition models as PSAMs for AR and GR with a 31-bp footprint, significantly larger than the 15-bp binding site core and even the 23-bp variable region of the SELEX probes. SelexGLM is capable of fitting such wide models because it considers offsets within the probe that partially cover the fixed sequences upstream of and downstream from the variable region. The corresponding energy logos for these PSAMs confirm the previously modeled 15-bp size of the GR binding site (Fig. 2B,D), but reveal AR's preference for poly(A) sequences flanking the 15-bp core (Fig. 2A,C) and surprising differences within the 3-bp central spacer (Fig. 2E; more evident in Supplemental Fig. S4C,G,H). For AR, replacing the best flanking sequence (AAAA) with the worst (TCGC) on one side leads to a 1.6-fold reduction in affinity (or 2.6-fold when both flanks are replaced) (Fig. 2E). Validation by qEMSA confirmed that spacer sequence affects the affinity of both AR and GR but that flanking sequences affect only AR (Fig. 2F). The observed change for AR (ΔΔG = 0.60 ± 0.05 kcal/mol) was larger than predicted (ΔΔG = 0.26 kcal/mol), perhaps because our model ignores dependencies between nucleotide positions within the binding site.
Further differences in AR and GR specificity are encoded throughout the 15-bp core
Though the AR and GR binding models are similar at first glance, detailed analysis of the PSAMs highlights AR's sensitivity to changes from the consensus sequence in the central 15-bp core region (Fig. 3A). Based on these differences, we tested sequences that favor AR (Fig. 3B; Supplemental Table S1, Shape-1 and Shape-2) and GR binding (Fig. 3B; Supplemental Table S1, Shape-4) and verified that the two proteins can differentiate between DNA sequences over an order of magnitude in affinity (Fig. 3B; Supplemental Fig. S7D). As the two proteins have identical base-reading chemistry at the DNA-binding interface, we examined whether their sequence preferences could be explained in terms of DNA shape readout. Indeed, the shape preferences of AR and GR contrast sharply: The AR benefits from a narrowed minor groove in the flanking regions (Fig. 3C), whereas the GR prefers binding to sites with widened minor grooves within the half-sites (Fig. 3D). To validate this finding, we tested a sequence with a half-site predicted to have a narrow minor groove (TTTTAT) (Zhou et al. 2013), and found that GR bound significantly worse than AR (Fig. 3B, Supplemental Fig. S7D, Shape-3). Similarly, we tested a site predicted to have a wide minor groove (GGGACA) (Fig. 3B; Supplemental Fig. S7D, Shape-4) and found a preference for GR. More dramatic examples of sequences predicted to have disparate GR and AR affinities are plentiful in the low-affinity range (KD ≥ 5 µM); however, measurements in this range are near the nonspecific binding limit and have not been reliable. In addition to differences in core preference, the nearly inverted minor groove preferences in the flanks for AR and the half-site for GR suggest the two proteins have different recognition modes.
Difference in DNA shape readout between AR and GR. (A) Difference in ΔΔG/RT values between AR and GR at each nucleotide position, normalized by their mean across all four bases. (B) Quantitative EMSA was used to measure the affinities of AR- and GR-DBDs for four sequences that maximally favor AR (Shape-1) and GR (Shape-4) and test the importance of half-site minor groove width (Shape-3 and -4). Each measurement was performed at least three times. Error bars, SEM. Average minor groove width profile for the top/middle/bottom 5% of sequences in terms of affinity for AR (C) and for GR (D).
The thermodynamics of binding reflect the specificity of AR and GR
To understand the thermodynamic basis of the differing AR and GR binding modes, we measured the effect of varying flanking and spacer sequences using isothermal titration calorimetry (ITC). Our ITC data allowed us to accurately quantify the affinity (and thus ΔG) of bound sequences and the contribution of individual sequence features to the enthalpy (ΔH) and entropy (ΔS) of binding (Buurma and Haq 2007). Although it was immediately apparent from the heat of binding (Fig. 4A) that AR and GR recognize the same DNA sequences differently, the KDs of AR and GR for DNA (Fig. 4B) were consistent with the PSAMs derived from the SELEX data (Supplemental Fig. S7C), including AR preference for poly(A) flanks. Overall, for the sequences tested, AR binding is more enthalpically driven (low ΔH) and GR binding more entropically driven (high TΔS) (Fig. 4C,D). This is consistent with previous findings; TF families engaged in direct readout via the major groove are more enthalpically driven, whereas those engaged in readout via the minor groove or backbone contacts are more entropically driven by solvent exclusion (Privalov et al. 2007, 2011). Our data also identify an exception to this general pattern: AR's recognition of the poly(A) stretches in the minor groove is driven not entropically but rather enthalpically. This observation contributes to our understanding of specificity: Enthalpic contributions to binding are not solely the result of hydrogen bonds with individual base pairs or backbone phosphates but can also result from interaction with a DNA feature; the narrowed minor groove (Fig. 3).
ITC analysis reveals distinct DNA-binding thermodynamics between AR- and GR-DBD. (A) The raw heat titration signals (top) and normalized heat of injection profiles (bottom) of AR- and GR-DBD bound to a given DNA sequence. Standard errors are estimated by NITPIC (Brautigam et al. 2016). (B) The KD of AR-DBD and GR-DBD for four sets of sequences fit from the ITC data. AR-DBD affinity is increased with flanking As and an optimal spacer, whereas GR is insensitive. (C) Enthalpy, ΔH, is calculated from the heat of binding for each DNA sequence. Flanking sequences decrease ΔH for AR-DBD, enhancing affinity. Smaller indicates a greater contribution to affinity. (D) Entropy, ΔS, is calculated from the KD (thus ΔG) and ΔH. GR-DBD affinity is more entropically driven. Larger indicates a greater contribution to affinity. (*) P-value ≤0.05, (**) P-value ≤0.01, (***) P-value ≤0.001, (****) P-value ≤0.0001, two-sided t-test. Error bars, SD represent the standard deviation from at least three experiments.
Promiscuous GR specificity predicts ability to bind at genomic AR loci
We next asked whether the SelexGLM models can explain why GR is able to functionally substitute for AR despite having nonoverlapping binding sites in models of CRPC.
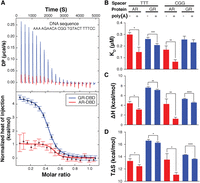
To this end, we analyzed ChIP-seq data from LNCaP-1F5, a cell line model of prostate cancer engineered to overexpress GR and enable genomic mapping of both AR and GR binding under similar cellular conditions (Sahu et al. 2013, 2014). As a positive control, we confirmed that the SelexGLM models can differentiate ChIP-seq peaks from adjacent regions (area under ROC-curve 0.78 and 0.83 for AR and GR, respectively) (Fig. 5A). Further, we observed a significant quantitative relationship between predicted affinity and degree of genomic occupancy (Fig. 5B), with ChIP-seq peaks in the top decile for in vitro affinity being significantly higher than those in the bottom decile (1.4-fold, P = 7.4 × 10−49, Wilcoxon rank-sum test, for AR; 1.6-fold, P = 2.8 × 10−68, for GR). To test whether the difference in intrinsic binding specificity between AR and GR discovered using SELEX-seq was reflected in the genomic occupancy patterns probed using ChIP-seq, we let our AR and GR PSAMs compete in a multiple linear regression model. As expected, when analyzing the variation in GR peak height in this manner, we found that the regression coefficient for GR affinity was significantly larger than that for AR (P < 10−6, t-test) (Fig. 5C) and that the latter did not deviate significantly from zero (P = 0.97). However, when analyzing the variation in AR peak height, both the AR and GR coefficients were nonzero (P < 10−5 and P < 10−7, respectively) and did not significantly differ from each other (P = 0.16), indicating that both AR and GR have the ability to bind well at AR loci.
Differences of intrinsic specificity between AR- and GR-DBD are reflected in the respective cellular genomic binding profiles. (A) Ability of the AR (red) and GR (blue) PSAMs to identify true ChIP-seq peaks as measured by the receiver operator characteristic (ROC) of binary classifiers identifying peaks versus adjacent regions. (B) Comparison of the median enrichment over background from ChIP-seq (y-axis) to the relative affinity for the strongest sequences within the ChIP-seq peak calculated from the position specific affinity matrix for AR (red) and GR (blue). In vitro affinity is binned by decile (e.g., 10 is the sequence space representing the top 10% highest affinity sites) (C) Multiple linear regression coefficients in models that use AR (red) and GR (blue) PSAM scores to predict AR (left) and GR (right) ChIP-seq peak enrichments. P-Values were calculated using t-tests.
Discussion
In this study, we developed and validated a strategy for inferring biophysical models of DNA-binding specificity of TFs from SELEX data. The computational method and software that we developed, SelexGLM, was instrumental for obtaining accurate estimates of the binding free-energy changes associated with any possible base substitutions in the DNA ligand over a footprint large enough to capture the flanking specificity outside the 15-bp core that we observed for AR. Experimentally, we designed a library to accommodate the footprint of AR and GR defined by crystal structures (Meijsing et al. 2009), isolated dimer-bound DNA from a mixed population by EMSA, and performed qPCR between rounds of enrichment to avoid PCR artifacts that add noise to measurements. These refinements revealed that, despite their similarities, AR and GR have differences in sequence specificity both within and outside the core 15-bp sequence that can be genetically tuned over a significant range, even in the absence of any cofactors.
HT-SELEX assays were previously performed on similar protein fragments (Jolma et al. 2013; Yang et al. 2017), and the resulting libraries were recently sequenced more deeply and then reanalyzed (Yang et al. 2017). Supplemental Figure S8 shows energy logos for binding models we inferred from these data using SelexGLM. A weak preference for poly(A) flanks outside the 15-bp core can be observed (Supplemental Fig. S8C), but it is not consistent between the three versions of the AR protein that was used, and the difference between AR and GR is not apparent. Yang et al. (2017) explicitly addressed the role of DNA shape readout, but their models for AR and GR (Supplemental Fig. S8E–G) did not cover the full footprint for either protein; therefore, no previous shape analysis for the flanking regions has been reported for AR or GR.
The intrinsic specificity, thermodynamics of binding, and genomic localization indicate that GR is more promiscuous than AR, allowing it to accommodate a wider range of sequences. These binding properties are consistent with the behavior of GR in some CRPCs. Anti-androgen therapy can result in de-repressed GR expression, increasing the amount of GR in the cell (Arora et al. 2013; Watson et al. 2015), which in the presence of endogenous or administered glucocorticoids results in an increase in the active concentration of GR in the nucleus. Our data are consistent with the idea that the more relaxed specificity provided by entropy-driven binding allows the excess GR to bind related sequences with reasonable affinity, including those previously bound by AR (Fig. 5C), enabling regulation of AR-driven genes aberrantly by GR (Arora et al. 2013). Thus, the biophysical properties of GR provide a rationale for how it can effectively substitute for AR in the context of CRPC.
The differences between AR and GR that our analysis uncovered were surprising, because structural analysis showed that all known AR and GR DNA contacts, and all amino acids within 6Å of the DNA, are identical. However, the integration of SELEX analysis and thermodynamic quantification of binding energies using ITC make it clear that AR and GR use these amino acids differently. Having comprehensively measured the specificity of AR and GR using all DNA ligands, it is clear that GR derives more entropic energy for the same DNA than AR. Because the majority of entropic energy is derived from solvent and ion exclusion from an interface and because the protein and DNA surfaces at the interface are identical in composition, we expected this contribution to be similar for AR and GR. However, the differences between GR and AR suggest an increase in conformational entropy within GR (Fig. 4D; Frederick et al. 2007), which would allow it to accommodate more diverse DNA sequences and shapes but decrease its specificity. AR, conversely, does not derive as much binding energy from entropy, suggesting that precise positioning of hydrogen bonding in the half-sites is more important. AR also derives enthalpic energy from the recognition of narrowed minor grooves created by poly(A) flanking sequences. Our results provide a thermodynamic basis for that discrimination. Our ITC data directly show that the increased negative electrostatic focusing (Rohs et al. 2009a) associated with a narrowed minor groove (Yoon et al. 1988) in the presence of interactions with basic residues (Supplemental Figs. S1, S2) drives an increase in affinity through enthalpy. These differences in the thermodynamics of recognition must be mediated by nonconserved amino acids within the fold of the protein, indicating a difference in how the interface is scaffolded. This phenomenon has also been observed in recent studies of the C2H2 Zinc finger family of TFs (Hughes 2011; Persikov et al. 2015) and indicates that both the residues displayed and how they are configured are critical to distinguishing how TF family members recognize different DNA sequences. It also suggests that, much as in the case of RUNX1 (Yan et al. 2004) and ETS1 (Pufall et al. 2005), the conformational entropy of GR may provide an opportunity for regulation that could direct it away from AR sites.
To analyze SELEX-seq data over a large enough footprint within an accurate biophysical modeling framework, we developed an algorithm named SelexGLM. In a self-consistent procedure, SelexGLM alternates between an “affinity-based alignment” step, which identifies the dominant binding site within each sequenced DNA ligand, and a GLM fitting step, which estimates the binding free-energy change associated with each possible base substitution in the DNA-binding site, until convergence is reached. The Poisson statistics that underlie the GLM fits allow us to obtain precise estimates of the free-energy parameters even when individual unique DNA have very few counts, since each possible base substitution or “feature” can occur in many different contexts. By using a modest sequencing depth of ∼107 reads per round, we were able to achieve precise, validated models over a footprint of 31 bp that would accommodate many other TF complexes.
SelexGLM also opens new avenues for studying the modulation of TF function. TF binding and activity are regulated by post-translational modifications (Pufall et al. 2005), cofactor binding (Chodankar et al. 2014), and combinatorial control through interaction with other TFs on DNA. The accurate modeling of binding specificity over an essentially unlimited footprint size by our method enables measurement of specificity for larger, heterologous complexes on DNA and the effect of sequence on their occupancy. Further, SelexGLM allows us to quantify relatively modest effects on specificity distributed over the entire footprint, which evolution may exploit to fine-tune expression levels in a flexible manner and which may be altered by phosphorylation of the TF or cofactor binding (Kumar and Calhoun 2008; Chodankar et al. 2014). Thus, our approach may enable us to begin to understand to what extent signal-dependent changes in expression are due to altered TF specificity.
Methods
Expression and purification of GR/AR-DBD
DNA sequences encoding the DBDs of human GR (GRα: 418–506) and AR (AR-B: 557–647) were cloned into an N-terminal his6-tagged vector (pET28a, Novagen). Vectors were transformed into BL21DE3 Gold Escherichia coli (Agilent) cells and grown to an OD600 of between 0.2 and 0.4. The temperature was reduced to 27°C, and 10 µM of ZnCl2 was added to the culture. Expression of recombinant protein was induced with 0.5 mM IPTG for 4 h when OD600 reaches 0.6 to 1. Cells were then spun down at 6000g for 15 min and then resuspended in Ni2+ column loading buffer (25 mM TrisHCl at pH 7.5, 500 mM NaCl, 15 mM imidazole, 1 mM DTT, 1 mM PMSF), snap frozen, and stored at −80°C until purification. Cell suspensions were lysed with an EmulsiFlex C3 homogenizer running at 15,000 psi. After three passes or more through the EmilsiFlex, soluble protein was isolated by ultracentrifugation at 40,000 rpm using a Beckman Ti-75 rotor for 1 h at 4°C and then collecting the supernatant. The supernatant was loaded onto nickel affinity column (GE Healthcare Life Sciences) pre-equilibrated with 25 mM TrisHCl (pH 7.5), 500 mM NaCl, and 15 mM imidazole. Unbound protein was washed off the column with equilibration buffer, followed by a low imidazole bump (25 mM TrisHCl at pH 7.5, 500 mM NaCl, 30 mM imidazole) to remove nonspecifically bound protein. A linear gradient of imidazole from 30 to 350 mM was used to elute DBD. Fractions containing DBDs were pooled and dialyzed overnight at 4°C in 20 mM TrisHCl (pH7.5), 50 mM NaCl, 2.5 mM CaCl2, and 1 mM DTT. Thrombin was used to cleave the his6-tag during the dialysis. Dialyzed protein was ultracentrifuged (40,000 rpm, 1 h, 4°C) and loaded onto a cation exchange column (HiTrap SP HP, GE Healthcare Life Sciences) pre-eliqulibrated with 20 mM TrisHCl (pH 7.5), 50 mM NaCl, and 1 mM DTT. The DBD was eluted in a linear gradient of NaCl from 50 to 350 mM over 20 CVs. Fractions containing DBD were pooled, concentrated (Amicon Ultra – 3K, Millipore), and filtered (Ultrafree-CL), and monomers were isolated by gel filtration (16/600 Superdex 200 PG, GE LifeSciences) in 20 mM HEPES (pH 7.7), 100 mM NaCl, 1 mM DTT. DBDs were collected and dialyzed against storage buffer (20 mM HEPES at pH 7.7, 100 mM NaCl, 1 mM DTT, 50% glycerol) and quantified (280 nm, ε = 5095M−1cm−1 for both AR- and GR-DBDs).
SELEX library design and synthesis
We designed our SELEX library to contain a 23-bp random region flanked by primer binding regions conforming to the Illumina TruSeq small RNA format for a total of 70 bp. The library was ordered in 1 mmol format from IDT as single strand with handmix option over the randomized region. The complementary strand was synthesized by Klenow extention using 5′-Cy5 labeled TSSR1 primer (Supplemental Table S1) on a PCR machine. Briefly, a reaction containing 2.5 µM ssDNA library, 5 µM 5′-Cy5-TSSR1, 150 µM dNTPs in NEB buffer 2 was incubated at 94°C for 3 min and then gradually cooled down to 37°C over 45 min. Six units of Klenow were added to every 25 µL of reaction, incubated at 37°C for 60 min, 72°C for 20 min, and gradually cooled down to 10°C over 45 min. dsDNA was purified using a MinElute PCR column (Qiagen) and quantified by absorbance at 260 nm.
SELEX-seq
Selection
We wished to achieve an average 1× coverage for all possible 23-mers. To this end, SELEX was carried out in a 120-µL binding reaction containing 0.63 µM purified DBD and 1 µM DNA library. At this size, there are ∼7.2 × 1013 DNA molecules in the reaction, representing ∼1.02× coverage of all possible 23-mers (7.03 × 1013). Binding was carried out in a buffer that approximates the salt and crowding of the nucleus for 1 h at 4°C (20 mM TrisHCl at pH 8.0, 150 mM KCl, 5% glycerol, 1 mM EDTA, 5 mM MgCl2, 40 ng/µL Poly(dIdC) [Sigma: P4925], 200 ng/µL BSA, 1 mM DTT, 200 mg/mL Ficoll PM400 [Sigma: F4375]). The reaction was then run out in multiple wells of a 10% native polyacrylamide gel (19:1 acrylamide/bis-acrylamide) in 0.5× TB containing 150 µM MgCl2 (89 mM Tris-boric acid, 150 µM MgCl2 at pH 8.3) at 4°C. In order to ensure that SHR-DBD:DNA complexes were trapped, the sample was loaded while running at 200 V to minimize the dissociation before entering the gel. DBD:DNA complexes were visualized using Cy5 fluorescence (GE ImageQuant LAS4010), isolated by excision, and bound DNA isolated by electroelution (Novagen D-tube dialyzer, 3.5 kDa) into a native PAGE running buffer as described above. Resulting DNA sequences were then purified using Qiagen MinElute PCR clean-up, eluted (10 mM TrisHCl at pH 8.0) to a final volume of 180 µL, and amplified to generate next round of library as described below. Because of the relatively low affinity of the SHRs for DNA, we were unable to shift enough DNA using a limiting amount of protein (<1:5) to allow PCR generation of pools for subsequent rounds without generating high-molecular-weight artifacts. We therefore incubated the library with a high protein:DNA ratio (0.63 µM:1 µM) to select all potential binding sequences in early rounds. Please note that this protein:DNA ratio requires more rounds of selection to begin linear enrichment of sequences. Since submission of this paper, we have begun using a 1:10 protein:DNA ratio. It is critical to perform a size-selection (8% 1× TG gel) from the recovered bound DNA to remove the high-molecular-weight DNA that comigrates with complexes prior to reamplification. Together with controlled amplification cycle by qPCR (see below), this improved SELEX protocol is artifact-free, saves a few rounds of selection, and is less saturated at early rounds. Typically, four to five rounds of selection are sufficient for factors with long binding footprints (∼25–30 bp).
Library regeneration
To generate enough DNA to perform each round of SELEX and sequencing library prep, the recovered DNA had to be carefully amplified. As these libraries were susceptible to amplification artifacts caused by overamplification (different type of artifact than those comigrating with the complex), the optimal number of PCR cycles was first determined by qPCR. Briefly, 1 µL of recovered dsDNA was analyzed in 50 µL qPCR reaction (0.5 µM TSSR0 primer [Supplemental Table S1], 0.5 µM unlabeled TSSR1 primer, 200 µM dNTPs, 0.1× SYBR green [Invitrogen: S-7563], 0.5 unit of Phusion polymerase in 1× NEB Phusion HF buffer). The amplification curve was then analyzed to determine the maximal number of rounds within the linear amplification range (typically less than 16 PCR cycles, depending on the amount of the template). Subsequently, 170 µL recovered library was divided into 85 reactions at 100 µL and amplified with the determined cycle numbers (with 5′-Cy5-TSSR1). The amplification reactions were then combined, purified, concentrated (Qiagen MinElute), and eluted with 45 µL EB. The resulting libraries were then quantified and mixed into a new binding reaction as described above. Additional agarose gel electrophoresis is required to remove poly(dIdC) if it is present in the recovered dsDNA, as the poly(dIdC) interfered with PCR.
Library sequencing
To sequence the resulting libraries on the Illumina HiSeq platform, additional adapter sequences were added by limited-cycle PCR. Briefly, 400 ng of each SELEX library was amplified using the 5′ adapter primer (TSSR2) (Supplemental Table S1) and the 3′ adapter and barcoding primer (TSSR-RPIX) (Supplemental Table S1) in a 1-mL PCR reaction (300 µM dNTPs, 0.8 µM TSSR2, 0.8 µM TSSR-RPIX, 10 U Phusion polymerase in 1× Phusion HF buffer) for two cycles. The added 71 bp allowed separation of the sequencing library from the adapter-less library on a 16% 0.5× TBE native polyacrylamide gel (19:1 acrylamide/bis-acrylamide) run at 200 V. The 141-bp band was excised and the DNA recovered by electroelution as described above. The purity and concentration of the library was determined by bioanalyzer (High-sensitivity dsDNA chip, Agilent). Multiple sequencing libraries with compatible barcodes were pooled in equimolar concentrations and sequenced on a HiSeq2000 with 10 million reads per library, using single-end, 50-bp sequencing mode.
Motif discovery
Raw sequencing data processing
The sequenced libraries were processed using the R package SELEX (R Core Team 2016; http://bioconductor.org/packages/SELEX). We required sequencing reads to match the sequence TGGAA at positions 24–28. The package was also used to construct Markov models of R0 and compute the information gain (KL divergence) after affinity-based selection.
Model-based analysis (SelexGLM)
To avoid bias in our estimates, we split the reads into two equal-sized random subsets. One half was used to define the “universe” of unique variable regions (which we refer to as “probes”). Read counts across this universe were defined based on the other half of the reads, and a count of zero was registered for probes that were only seen in the first half. A Markov model of order 5 was constructed from the R0 probes using the selex.mm() function from the SELEX package, and an affinity table for k = 15 was constructed using selex.affinities(). An initial PSAM was constructed from the relative affinity of all 15 × 3 single-base mutations of the optimal 15-mer, expanded to the desired size by adding eight neutral columns on each side, and used as a seed. The subsequent iterative procedure alternated between two steps. First, the current PSAM was used to find the position/direction of highest affinity on either strand, the optimal “view” on the probe. If that optimal affinity was >95% of the sum over all positions (including the top position), the probe was used in the analysis; otherwise, it was ignored. The set of optimal positions in each of the accepted probes was used to define a design matrix containing the base identity at each position relative to the start of the optimal view. By using the probe counts as independent variables, the logarithm of the expected probe frequency in R0 according to the Markov model as offset, and a logarithmic link function, a fit was performed using the glm() function. The regression coefficients were interpreted as free-energy differences ΔΔG. All our analyses were implemented in the form of an R software package, deposited in Bioconductor (R Core Team 2016; https://www.bioconductor.org/). All computational figure panels in this paper were produced fully automatically from the raw sequencing data using R scripts that use the SELEX and SelexGLM packages.
ITC
ITC was performed using a Microcal VP-ITC (GE) at 25°C. Protein and DNA samples were dialyzed into binding buffer (20 mM HEPES-KOH at pH 7.7, 250 mM KCl, and 0.5 mM TCEP) at 4°C for 36–48 h before use. DNA samples were loaded into the syringe and titrated into the protein in the reaction cell. Each ITC experiment consisted of an initial 2 µL injection, followed by 20 × 14.3 µL injections, with 240 sec between injections. For AR-DBD, 50 µM DNA and 10 µM protein sample were used. For GR-DBD, 100 µM DNA and 20 µM protein sample were used to increase signal. The raw isotherm was analyzed using NITPIC (Brautigam et al. 2016), followed by fitting to a one-site binding model in SEDPHAT (Brautigam et al. 2016). The mean and standard deviation of thermodynamic parameters were calculated based on at least three experimental replicates.
ChIP-seq data analysis
Raw reads for AR (GSM759657 and GSM759658), GR (GSM759669), and IgG control (GSM759671) data from ChIP-seq experiments using LNCaP-1F5 cells were downloaded from Gene Expression Omnibus and aligned to the hg19 assembly using Bowtie 2 (Langmead and Salzberg 2012) with settings “--sensitive --score-min L,-1.5,-0.3.” Peaks were called using MACS2 (Zhang et al. 2008; https://github.com/taoliu/MACS) width default settings, and the “fold enrichment” (column 7) was used to quantify peak strength. The narrowPeak bed-file was filtered to retain one entry per unique peak interval, and the intervals was standardized to cover 200 bp. The PSAM score was then calculated for each offset and orientation of the peak, and the largest value was recorded.
Statistical analysis
Figure 1
To minimize the influence of sequencing count on the calculation of relative enrichment, we only used 15-mers with 100 or more counts in both the R7 and R8 libraries. We consider the sequencing count of each k-mer as a random variable of Poisson distribution, where the k-mer count (n) is the best estimation of the mean (λ). Therefore, the variance is approximated by the k-mer count, with an absolute error of Sqrt (n). A sequencing count of 100 or more therefore restricts the absolute error no more than 10% of the mean (λ).
Figures 2F, 3B, 4, Supplemental Figures S4, S7
To compare DNA-binding affinities of GR and AR for different sequence by EMSA or ITC, we performed at least three independent experiments under the same conditions. The variance of measured affinity is assumed to be normally distributed, and the t-test and P-values are appropriate.
Data access
The raw SELEX-seq reads from this study have been submitted to the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Leinonen et al. 2011) under accession number SRP101815. SelexGLM has been deposited in R/Bioconductor (https://www.bioconductor.org/). Scripts for running SelexGLM on our data sets and HT-SELEX data are included as Supplemental data.
Acknowledgments
We thank Todd Riley and members of the Pufall, Bussemaker, Mann, and Rohs laboratories for useful discussions. This research was supported by the National Institutes of Health (NIH) K99/R00 CA149088, the Roy J. Carver Charitable Trust 01-224, and National Science Foundation CAREER grants 1552862 (M.A.P.) and R01HG003008 (NIH) (H.J.B.).
Author contributions: M.A.P. and H.J.B. conceived and oversaw the project. L.Z. designed and performed all experiments. G.D.M., J.F.K., C.R., and H.J.B. conceived and developed the GLM analysis approach. G.D.M. implemented the SelexGLM software and analyzed SELEX-seq data. H.T.R. analyzed ChIP-seq data. L.Z. and V.D.F. performed preliminary analysis of SELEX-seq and ChIP-seq data. M.A.P. analyzed the crystallography data. J.C.H. provided the ITC hardware and consultation on interpretation. L.Z., G.D.M., H.T.R., H.J.B., and M.A.P. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.222844.117.
- Received March 14, 2017.
- Accepted November 22, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








