
RNA structure replaces the need for U2AF2 in splicing
- Chien-Ling Lin1,4,
- Allison J. Taggart1,4,
- Kian Huat Lim1,4,
- Kamil J. Cygan1,2,
- Luciana Ferraris1,
- Robbert Creton1,
- Yen-Tsung Huang3 and
- William G. Fairbrother1,2
- 1Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, Rhode Island 02912, USA;
- 2Center for Computational Molecular Biology, Brown University, Providence, Rhode Island 02912, USA;
- 3Departments of Epidemiology and Biostatistics, Brown University, Providence, Rhode Island 02912, USA
- Corresponding author: fairbrother{at}brown.edu
-
↵4 These authors contributed equally to this work.
Abstract
RNA secondary structure plays an integral role in catalytic, ribosomal, small nuclear, micro, and transfer RNAs. Discovering a prevalent role for secondary structure in pre-mRNAs has proven more elusive. By utilizing a variety of computational and biochemical approaches, we present evidence for a class of nuclear introns that relies upon secondary structure for correct splicing. These introns are defined by simple repeat expansions of complementary AC and GT dimers that co-occur at opposite boundaries of an intron to form a bridging structure that enforces correct splice site pairing. Remarkably, this class of introns does not require U2AF2, a core component of the spliceosome, for its processing. Phylogenetic analysis suggests that this mechanism was present in the ancestral vertebrate lineage prior to the divergence of tetrapods from teleosts. While largely lost from land dwelling vertebrates, this class of introns is found in 10% of all zebrafish genes.
RNA splicing is a process that removes an internal segment of RNA (i.e., the intron) and rejoins together the two flanking segments (exons). Distinct but evolutionarily related versions of this processing reaction are found in prokaryotes and eukaryotes in a variety of different contexts. In eukaryotes, the splicing of nuclear introns is catalyzed by a large riboprotein complex called the spliceosome (Matlin and Moore 2007). RNA encoded by genes in organelles and some bacterial genomes contain self-splicing group I and II introns which catalyze their own removal (Cech et al. 1981). A basic problem for all introns is the correct identification and pairing of the splice sites. In group I and II introns, this pairing function is performed by RNA secondary structure alone, whereas in spliceosomal introns, small nuclear ribonucleoproteins (snRNPs) recognize and pair together the correct 5′ splice site (5′ ss) and branchpoint site (BP). However, there are some examples where the pairing of sites is assisted by intramolecular secondary structure in the intron (Goguel and Rosbash 1993; Libri et al. 1995; Charpentier and Rosbash 1996; Howe and Ares 1997; Spingola et al. 1999). In addition, there are some fascinating examples of how secondary structures can regulate mutually exclusive alternative splicing (Warf and Berglund 2007; McManus and Graveley 2011): Several regions of the Dscam1 pre-mRNA undergo extensive alternative splicing. In one of these regions, an upstream “selector” sequence near exon 5 can select from an array of 48 complementary downstream “docking” sequences. Each “docking” sequence can potentially base-pair with the “selector” sequence, thereby bringing an alternate version of exon 6 to splice to exon 5 (Celotto and Graveley 2001; Graveley et al. 2004; Graveley 2005; Kreahling and Graveley 2005; May et al. 2011). As only a single hairpin can form, only a single 3′ splice site (3′ ss) can pair. Recent work suggests analogous mechanisms may explain regulated splicing at several other loci (Yang et al. 2011).
Secondary structure in RNA can be identified experimentally or computationally. There are currently around a thousand publicly available structures—53% determined by X-ray crystallography and 47% by solution NMR (Bernstein et al. 1977). There have been a great many advances in computational approaches to predicting secondary structures (Mathews 2006; Mathews et al. 2007; Seetin and Mathews 2012). A variety of algorithms are currently in use, the most common being free energy minimization, which are increasingly used in combination with comparative sequence analysis and protection/enzymatic mapping approaches (Mathews 2006; Bellamy-Royds and Turcotte 2007; Low and Weeks 2010). A functional role for a predicted secondary structure has typically been explored by a two-step process of introducing mutations to disrupt predicted structure, followed by compensatory mutations at a second site designed to restore structure (Chen and Stephan 2003).
Here, we report a functional role for expansions of simple repeats that is mediated by RNA secondary structure. These simple repeats were discovered using a computational method for detecting rapidly evolving noncoding splicing elements (Lim et al. 2011). A combination of chemical mapping of RNA structure, compensatory mutation analysis, and in silico RNA folding was utilized to define a novel class of structured introns.
Results
Intronic repeats of AC and GT are fish-specific splicing elements
As a splicing element's function often depends on its location, natural selection results in the accumulation of functional cis-elements at optimal positions relative to active splice sites (Lim et al. 2011). We reasoned that, since the positional distribution of a motif relative to splice sites appears to be a signature of its function, a difference in these signatures across species may represent a change in the role of a splicing element in at least one of the lineages in the comparison. In contrast, a motif with a similar positional distribution in two species is probably performing a similar function in both species. For example, a sequence that is involved in a conserved process like the basic catalysis of splicing (e.g., 5′ ss or branchpoint motif) has a similar distribution in zebrafish and human (Fig. 1A,C). The sequence elements with the greatest difference observed across six pairwise comparisons between human and six other vertebrates were the simple dinucleotide AC and GT repeats such as ACACAC, CACACA, GTGTGT, and TGTGTG (Fig. 1B,C; Supplemental Fig. 1; see Methods for a description of the L1 distance metric). This difference was largely accounted for by the presence of AC and GT repeats in zebrafish introns. Further analysis of other available fish and lamprey genomes suggests AC and GT repeats are potentially performing some sort of lineage-restricted role in splicing (Supplemental Table 1; Supplemental Figs. 2, 3).
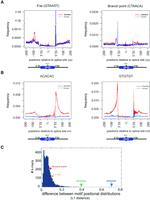
The distribution of AC and GT repeats shows the most extreme divergence between human and zebrafish. (A) Plot of frequency (y-axis) of 5′ ss and BP around all 3′ and 5′ ss in human (blue line) and zebrafish (red). The position plotted is −200 to +100 relative to 3′ ss (−200 to “//” of x-axis) and −100 to +200 relative to 5′ ss (“//” to 200 of x-axis). (B) Plot of the frequency (y-axis) of AC and GT repeat hexamers around all 3′ and 5′ ss in human (blue line) and zebrafish (red). (C) Differences of motif positional distributions (L1 distance) of all 4096 hexamers in human-zebrafish comparison. The distance is the sum of the difference (e.g., area between two lines in A and B) of normalized frequencies relative to splice sites. Green arrows show AC repeat hexamers (i.e., ACACAC and CACACA). Blue arrow shows GT repeat hexamers. Yellow and red arrows indicate 5′ ss and BP, respectively.
Upstream repeats of AC co-occur with GT repeats across introns but not exons
To better understand the differences in distribution profiles between AC and GT repeats in fish versus other vertebrates, we examined the occurrence frequency of the hexamers ACACAC and GTGTGT around splice sites in zebrafish and human genomes. The most obvious feature in the comparisons were prominent peaks of AC repeats downstream from the 5′ ss and GT repeats immediately upstream of the 3′ ss in zebrafish (Fig. 1B). As runs of AC and GT repeats are complementary, these intronic elements could base-pair with each other provided they co-occurred either (1) across the exon causing exon looping, or (2) across the intron causing an intron bridging structure to form. To test these two models, we compared the observed frequency of co-occurring (AC)m-(GT)n repeats against a null model of uniform AC and GT distributions. As it was not clear what length of dinucleotide repeats is necessary for robust hairpin formation, we analyzed this data at several different repeat lengths. In each case, we calculate a value for the co-enrichment AC and GT repeats above background. Plotting these data as a heat map demonstrates a higher enrichment of co-occurrence across introns than exons (Fig. 2). There are very few runs of intronic AC repeats adjacent to downstream runs of intronic GT repeats across the exon (absence of signal in center of heat map) (Fig. 2, right). In contrast, large runs of AC repeats tend to co-occur with large runs of GT repeats across introns, implying a selection for pairings of longer tracts of AC and GT repeats in zebrafish introns (presence of signal in center of heat map) (Fig. 2, left). This distribution is consistent with AC and GT repeats forming an intron-bridging secondary structure.
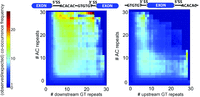
AC and GT repeats co-occur across introns, not exons. Zebrafish introns containing AC repeats within 200 nt of the 5′ ss were examined for downstream intron GT repeats (left panel) or upstream intron repeats (right panel). Introns were binned according to the length of the GT repeats (# of repeats, x-axis) and AC repeats (# of repeats, y-axis). The observed frequency of introns with each combination of repeat length is shown on the graph. The heat map color of each bin indicates the fold difference of observed frequency over the co-occurrence frequency predicted by a model of independently distributed AC and GT repeats.
AC and GT repeats are predicted to mediate highly stable structures that form across the intron
To explore the idea that AC and GT repeats are associated with increased secondary structure across zebrafish introns, we
used the RNA structure prediction program, RNAfold (Zuker and Stiegler 1981), to predict the degree of secondary structure in zebrafish introns. Introns that contain at least one occurrence of both
the AC and GT hexamers at the 5′ and 3′ intron termini have a striking 1.5- to twofold lower ΔG across a range of intron lengths
(Methods; Fig. 3A) (P value < 0.001). Examining the distribution of predicted stabilities of all introns in this size range demonstrates that (AC)m-(GT)n introns form a separate class of structured introns (Fig. 3B). It is possible that this elevated stability is due to the co-occurrence of complementary dinucleotides or the entire intron
is under selection to fold into a stable secondary structure. To test if this increased stability is due to composition alone,
(AC)m-(GT)n introns were shuffled in a manner that preserved dinucleotide frequencies and refolded. On average, native introns were a
third more stable than shuffled controls 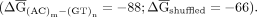 This analysis suggests a class of introns whose sequence is under selection to form stable secondary structure that is mediated
by complementary runs of AC and GT repeats. We hypothesized that this predicted structure both forms and is necessary for
accurate splice site pairing. To test this hypothesis, we selected an exemplar intron that contained AC and GT repeats (intron
5 of cep97 [centrosomal protein of 97 kDa]) for detailed analysis. This exemplar, typical of this class of introns (Table 1), contains extensive uninterrupted runs of AC and GT repeats (i.e., >14 repeats) that obscure signals such as BP and polypyrimidine
tract (PPT) at the 3′ end of the intron. The predicted structure includes >75% of all intronic positions in a double-stranded
state of extremely low energy (ΔG = −99.7 kcal/mol, 99th percentile in 170-nt bin) (Fig. 3B). A plot of the predicted structure suggests that base-pairing between the AC and GT repeats forms a large hairpin that
brings the 3′ ss and 5′ ss into close proximity (Fig. 3C).
This analysis suggests a class of introns whose sequence is under selection to form stable secondary structure that is mediated
by complementary runs of AC and GT repeats. We hypothesized that this predicted structure both forms and is necessary for
accurate splice site pairing. To test this hypothesis, we selected an exemplar intron that contained AC and GT repeats (intron
5 of cep97 [centrosomal protein of 97 kDa]) for detailed analysis. This exemplar, typical of this class of introns (Table 1), contains extensive uninterrupted runs of AC and GT repeats (i.e., >14 repeats) that obscure signals such as BP and polypyrimidine
tract (PPT) at the 3′ end of the intron. The predicted structure includes >75% of all intronic positions in a double-stranded
state of extremely low energy (ΔG = −99.7 kcal/mol, 99th percentile in 170-nt bin) (Fig. 3B). A plot of the predicted structure suggests that base-pairing between the AC and GT repeats forms a large hairpin that
brings the 3′ ss and 5′ ss into close proximity (Fig. 3C).
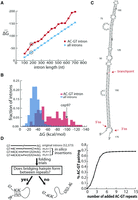
(AC)m-(GT)n introns represent a separate, structured class of introns in zebrafish. (A) Zebrafish introns are binned by length and folded by RNAfold. The average minimum free energy of folding for each bin is plotted for all introns and the subset of (AC)m-(GT)n introns defined by at least one occurrence of an AC and a GT repeat hexamer within 40 nt of the 5′ ss and 3′ ss, respectively. The 170-nt bin (circled) is expanded in B to show the distribution of the free energies of intron folding for the general population of introns (blue) and the (AC)m-(GT)n subclass (red). (C) The structure of an exemplar intron (intron 5 from cep97) was predicted and displayed by RNAfold. The 5′ ss, the 3′ ss, and the branchpoint site (determined experimentally by inverse PCR in vivo) are indicated with red arrows and text. (D) (AC)m-(GT)n repeat addition simulations in zebrafish introns demonstrating the effect of AC and GT repeats on intronic structure. (Left) Schematic for (AC)m-(GT)n repeat addition simulation. AC and GT dinucleotide repeats of varying length (0 ≤ m, n ≤ 20) were added 20 nt downstream from the 5′ ss and upstream of the 3′ ss. The repeats were determined to direct the overall structure if the upstream AC repeats base-paired to the downstream GT repeats forming a large hairpin. (Right) Simulation result. The percentage of introns in which the AC repeats base-paired to the GT repeats to form a hairpin structure was counted (y-axis) against the insertion of varying lengths of AC and GT repeats (x-axis). Resampling the data resulted in 95% confidence intervals of <1%.
Examples of AC-GT introns in zebrafish
While the selected example contains a striking amount of intron bridging structure, it was not clear whether shorter runs of AC and GT repeats could also create bridging interactions. To determine the minimal number of repeats that are required to drive intron bridging structure, a simulation approach was applied to 52,373 zebrafish introns that lacked AC and GT repeats. Briefly, short blocks of (AC)m and (GT)n repeats with incrementally increasing values of m and n were inserted in silico to the 5′ and 3′ ends of the intronic sequences prior to RNA structure prediction. The repeats were judged to drive structure if the AC repeat insert was paired with the GT repeat insert. Structure prediction results demonstrate that a repeat length of six was sufficient to create a maximum amount of intron bridging (see asymptote at maximal 66% value) (Fig. 3D). This effect is independent of intron length (Supplemental Fig. 4). The structural analysis suggests that co-occurring (AC)m-(GT)n repeats bridge the introns between splice sites and stabilize their secondary structures.
AC and GT repeats are required for accurate 5′ ss and 3′ ss pairing in vivo
To ensure that these repeat expansions were functional and not background mutations, we assayed the splicing of the cep97 exemplar intron and a control intron that contained no AC or GT hexamer repeats (intron 16 of sacm1lb [SAC1 suppressor of actin mutations 1-like b]) in zebrafish. Both cep97 and the control intron spliced constitutively, demonstrating that highly structured introns with poor matches to consensus splice signals can splice efficiently in vivo (Fig. 4A,B). The hypothesis that this class of zebrafish introns requires secondary structure for correct splicing was tested in two steps—(1) enzymatic mapping was used to probe structure and (2) a mutational approach was used to test function. Since the extensive mutations that are required to disrupt this structure may have the unintended consequence of destroying splicing elements, we substituted each strand of the hairpin sequence with the corresponding region of the control intron. The constructs (AC-GT, CON-TROL) and the resulting chimeras (AC-TROL, CON-GT) were tested in two different minigenes (see design in Fig. 4A). The control intron spliced correctly in the reporter as did the AC-TROL chimera (Fig. 4B, lanes 3,4). However, the chimera CON-GT does not splice accurately (Fig. 4B, lane 5). In addition to the correct product, the chimeric minigene's transcript is spliced to a cryptic 3′ ss in the downstream exon. This poor specificity of 3′ ss usage is rescued by the re-introduction of AC repeats, which reconstitutes the endogenous intron (Fig. 4B, lane 6). These results demonstrate that, while the control splice sites are utilized in all contexts, the 3′ ss downstream from the GT repeats requires pairing with the upstream AC repeat for accurate 3′ ss utilization.
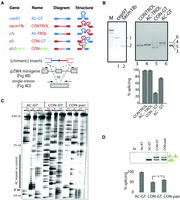
The predicted hairpin in (AC)m-(GT)n introns is required for accurate splice site recognition. (A) The exemplar (AC)m-(GT)n intron from zebrafish cep97 intron 5 (blue) and a control from sacm1lb intron 16 (red) were recombined at their midpoints and cloned into splicing reporters to make chimeric intronic constructs. Construct nomenclature derives from suffix/prefix combinations listed on the left. Green color indicates regions of complementarity used to make compensatory mutations in CON-pair constructs. (B) RT-PCR from total RNA extracted from whole-body juvenile zebrafish. Primer amplicons span four exons centered on the AC-GT or CONTROL intron. RT-PCR from total RNA extracted from transiently transfected tissue culture cells and quantified (histogram). The predicted sizes of the constitutively spliced products are illustrated with arrows marked by lane numbers. (C) RNase mapping of AC-GT, CON-GT, and CON-pair introns. Structured regions inferred from protection from single-strand nucleases (see RNases T1 and A). (D) Intron substrates used in mapping were tested in a single intron splicing reporter construct, transfected into cells, and assayed and quantified as described above. Asterisk indicates statistical significance by paired t-test (P = 9 × 10−4) of three biological replicates.
To test if this requirement depends on the formation of secondary structure, we employed RNase mapping to confirm the predicted double-stranded structure forming between AC and GT repeats (Fig. 4C). Three RNAs were transcribed in vitro for the RNase mapping assay: AC-GT and CON-GT introns as described in Figure 4A, and the CON-pair intron of which the last 35 nt were mutated to base-pair with the first 35 nt of the “CON” sequence. Shown by serial titrated RNase digestion, the AC repeats in the 5′ intron region of the AC-GT intron were protected from single-stranded nucleases (Fig. 4C, “AC-GT”). This protection was lost when the 5′ intron was substituted with sequences that could not pair with the downstream GT repeats (Fig. 4C, “CON-GT”) but could be partially restored by compensatory mutations in the 3′ intron that restored predicted bridging structure (Fig. 4C, “CON-pair”). Assaying the splicing of these introns in a single intron minigene (depicted in Fig. 4A) confirms the dependence of the GT repeat-rich 3′ region on upstream runs of AC repeats and also demonstrates that compensatory mutations that re-introduce intron-bridging structure partially restore the splicing (Fig. 4D). The compensatory mutations predicted to restore structure eliminated the BP used in the wild-type intron, which may explain the limited rescue (Fig. 3C). Taken together, these results demonstrate an intron bridging structure forming between upstream AC repeats and downstream GT repeats that is necessary for accurate 3′ ss usage in vivo.
The splicing of (AC)m-(GT)n introns does not require basal splicing factor, U2AF2
To further define the biochemical requirements of splicing (AC)m-(GT)n introns, the single intron substrate (Fig. 4C) was used to establish an in vitro splicing assay. Here, pre-mRNA substrate of zebrafish cep97 (AC-GT) and the adenoviral positive control (Ad81) were transcribed in vitro and incubated in HeLa nuclear extract. Distinct species corresponding to the known intermediates and products of the two-step splicing reaction, namely lariat intermediate, lariat, free 5′ exon, and ligated exons, can be visualized on a denaturing gel (Fig. 5). Prior studies have demonstrated that in vitro splicing is particularly sensitive to even short stretches of secondary structure (Solnick and Lee 1987). However, the naturally structured AC-GT intron spliced nearly as efficiently as the adenovirus positive control (Fig. 5). To probe the protein requirements of the (AC)m-(GT)n splicing reaction, we systematically targeted basal spliceosome components associated with the bridging of the 5′ ss to 3′ ss that occurs early in spliceosome assembly with blocking antibodies. While irrelevant antibodies did not inhibit splicing, we found that pre-incubation of extract with anti-U2AF2 antibodies inhibited the control Ad81 intron but not the AC-GT intron (Fig. 5A). This antibody has previously been demonstrated to block U2AF2 interactions with substrate (Gama-Carvalho et al. 1997). U2AF2 is a single-stranded RNA binding protein that recognizes the PPT upstream of the 3′ ss and recruits U2 snRNP to the BP. Applying a scoring algorithm for PPT suggests 61% of (AC)m-(GT)n introns lack PPT in zebrafish (Supplemental Fig. 5). Pre-incubation with antibodies against SF3B1, SR proteins and o-methyl oligonucleotides targeting U1snRNP affected both the control and (AC)m-(GT)n intron equally, suggesting these factors were required by both types of introns (Fig. 5B; Supplemental Fig. 6). These experiments and the lack of an obvious PPT (Table 1) suggest (AC)m-(GT)n introns diverge from typical introns in the early recognition steps at the 3′ ss.
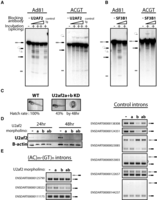
The splicing of (AC)m-(GT)n introns requires components of U2 snRNP but not U2AF2. In vitro splicing substrates were prepared from the constructs used in Figure 4 and the model in vitro splicing construct Ad81. The in vitro–transcribed RNA was incubated in HeLa nuclear extract for the splicing assay. (A) The splicing of the Ad81 control was compared to the (AC)m-(GT)n intron with pre-incubation with either: no antibody, a control antibody, or increasing amounts of anti-U2AF2 antibody. The input pre-mRNA and the splicing products, including the lariat intermediate and free first exon from the first step of splicing, the free lariat, and ligated exon from the second step of splicing, were resolved on an 8M urea gel and visualized by autoradiography with a phosphoimager. (B) The comparison described above was repeated with antibody targeting SF3B1, a component of the U2 snRNP. (C) u2af2 knockdown (KD) in zebrafish embryos. The KD embryos developed without gross phenotypic defect by 48 h but with lower hatch rate. (D) Western blotting of U2af2 24 or 48 h after injection. Beta actin served as a loading control. (E) RT-PCR of single intron transcripts containing (AC)m-(GT)n repeats (left) or without the repeats (right). Pre-mRNA and/or spliced mRNA is depicted on the right of the gel images. A bracket indicates the smear of the PCR product, possibly due to the loss of splicing accuracy.
To test the U2af2 requirement of (AC)m-(GT)n intron splicing in vivo, morpholino oligo injection was used to block the translation of U2AF2 paralogs in zebrafish embryos. Zebrafish carries two U2af2 homologs, U2af2a and U2af2b. These paralogs share 88% sequence identity; they both encode three RNA-recognition motifs (RRMs), a U2AF interacting domain, and an arginine and serine-rich (RS) domain, in which resides the most sequence variation between two genes (Supplemental Fig. 7). As U2AF2 RS domains are involved in pre-mRNA binding and protein-protein interactions (Valcarcel et al. 1996; Rudner et al. 1998a,b), U2af2a and 2b may have distinct pools of RNA substrates and/or interacting proteins. While the depletion was efficient, the phenotype was modest (Fig. 5D). U2af2-depleted embryos developed into fish with a lower hatch rate compared to their wild-type controls (control: 100%, u2af2a KD: 47%, u2af2b KD: 47%, u2af2a+2b KD: 43%) (Fig. 5C,D). While RNA-seq did not reveal significant differences in splicing, most AC-GT introns coexist with non-AC-GT introns in large multi-intron transcripts and therefore could not be studied in isolation. To study the effects of U2af2 depletion in isolation, nine single intron genes were analyzed by RT-PCR. The genes containing AC-GT introns were unaffected by U2af2a, U2af2b, or U2af2a+b depletion (Fig. 5E, left). However, half of the genes containing a non-AC-GT intron exhibit a defect in splicing in at least one of the depletion experiments (Fig. 5E, right) Taken together, both in vitro and in vivo assays demonstrate that U2af2 is not required for the processing of (AC)m-(GT)n introns.
AC and GT repeats appear to be an ancient splicing mechanism that preceded the divergence of tetrapods from fish
With the availability of completely sequenced genomes, the analysis described in this study can be readily extended across multiple species of vertebrates. Analyzing 24 genomes, we find the association of AC with the 5′ ss intronic flanking sequences and of GT with the 3′ ss mostly restricted to teleosts, the ray-finned bony fish (Fig. 6). The genomic signature is most prevalent in zebrafish. Interestingly, this type of intron is also present in the lamprey, whose last common ancestor preceded the evolution of jawed vertebrates and the divergence of tetrapods from fish, which suggests AC and GT concurrent repeats to be an ancient splicing mechanism. The proportion of all introns defined as (AC)m-(GT)n introns ranged between 0.09% and 2.29% across fish species (Supplemental Table 1). In zebrafish, 10% of all genes contain an (AC)m-(GT)n intron. (AC)m-(GT)n introns are observed in both constitutive and alternatively spliced introns with similar frequencies [2589 (AC)m-(GT)n introns in 98,694 total detected introns, 53 (AC)m-(GT)n introns in 1756 alternatively spliced introns, and 1 (AC)m-(GT)n intron in 22 significant differential splicing events comparing zebrafish adult head and tail tissues].
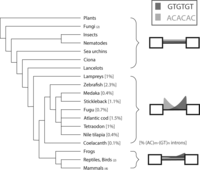
(AC)m-(GT)n introns predate the divergence of tetrapods from teleosts. The measurement of frequency distribution of AC and GT repeat hexamers around 5′ ss and 3′ ss was expanded to 24 genomes (listed in dendrogram). The degree and shape of AC and GT enrichment is illustrated in the cartoon. The percentage of (AC)m-(GT)n intron in the genome is indicated in brackets, and the number of genomes analyzed in the category is in parentheses.
Existing models of splice site recognition argue that mechanisms of intron definition [like the (AC)m-(GT)n pairing] tend to occur in shorter introns (Robberson et al. 1990). However, comparing the length of (AC)m-(GT)n introns to the genome-wide average intron length across several species suggests (AC)m-(GT)n intron lengths regress toward the mean (Supplemental Table 1; Supplemental Fig. 8). In other words, (AC)m-(GT)n introns tend to be larger than average in fish with short introns (i.e., fugu, tetraodon, cod, stickleback, medaka, and zebrafish, with median intron length <1 kb) and shorter than average introns in fish species with larger introns (i.e., coelacanth and tilapia, with median intron length 1–2 kb) (see Supplemental Table 1 for P value from genome-wide comparison). Moreover, the length variance within (AC)m-(GT)n introns is significantly smaller than those without (AC)m-(GT)n repeats in seven out of nine fish genomes examined (Supplemental Table 1). These data argue that (AC)m-(GT)n introns evolved toward an optimal range of length within and across species due to their structural constraints.
As the phylogeny suggests that (AC)m-(GT)n repeats were lost in humans, we examined the set of human orthologs to zebrafish (AC)m-(GT)n introns to discover other splicing mechanisms that may have arisen to compensate for the loss of (AC)m-(GT)n pairing (Supplemental Table 2). The most obvious changes were found at the 3′ ss region. The human counterparts of (AC)m-(GT)n introns had extended PPT and more purine rich exonic splicing enhancer (ESE)-like elements in their downstream exons (Supplemental Fig. 9). As U2AF2 binds the PPT, this observation further supports compensatory roles of bridging structure and U2AF2 binding in splicing. Furthermore, there appears to be less reliance on the U2AF2 in fish than in humans. The depletion of U2af2 does not appear to affect cell viability or organismal morphology, and significantly more fish introns lack a PPT at the 3′ ss (23% in fish versus 16% in human) (Fig. 5; Supplemental Fig. 5).
Computational analysis suggests G triplets may contribute to intron bridging structure in humans
Finally, we address the question of whether other types of modular structured introns enforce splice site selection in other
species. Human introns were folded and stratified according to stability (i.e., ranked by ΔG). To identify other motifs that
define new subclasses of introns in humans, we focused on highly structured introns and measured co-occurrence frequency of
complementary k-mer pairs across the intron (Methods). Many structured introns (16%) had three or more G triplets in the first 40 nt of the
intron, followed by three or more instances of the complementing C triplets in the last 40 nt. This arrangement of previously
identified G triplet splicing enhancer signals occurs four times more frequently than expected by chance (P value < 0.0005) (McCullough and Berget 2000). Introns with this arrangement of elements are significantly more stable (P value < 0.001) than their shuffled controls; however, this effect is not nearly as striking as their zebrafish counterparts
(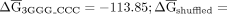 −111.47 —controlled to preserve GGG and CCC frequency). Overall, this analysis suggests that a variety of combinations of
G-, C-, and GC-rich repeats (Supplemental Table 3) could be playing structural roles in pairing splice sites across mammalian
introns.
−111.47 —controlled to preserve GGG and CCC frequency). Overall, this analysis suggests that a variety of combinations of
G-, C-, and GC-rich repeats (Supplemental Table 3) could be playing structural roles in pairing splice sites across mammalian
introns.
Discussion
Here, we report a class of introns that requires secondary structure for correct processing (see model in Fig. 7). This analysis started as a pairwise comparison between human and a series of six other vertebrate species to identify rapidly evolving splicing elements (i.e., lineage-specific splicing elements) (Supplemental Fig. 1). In each of the pairwise comparisons, the analysis was restricted to orthologous exon regions, which means that any differences observed arose within existing genes and are unaffected by gene duplication or acquisition in one lineage. The most striking difference observed between human and any other vertebrate is a shift in the distribution pattern of AC and GT repeats seen in the regions around splice sites in a fish-to-human comparison (Fig. 1). While these AC and GT repeat motifs had been previously reported to be specifically enriched in fish introns with weak splice sites by pan-species computational screens (Yeo et al. 2004), we demonstrate that these elements, found within 200-nt intronic windows adjacent to the 5′ and 3′ splice sites, co-occur across introns but not across exons, and work together to enforce accurate splice site pairing (Figs. 2⇑–4).
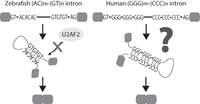
Model for secondary structure-dependent splicing. (Left) Zebrafish (AC)m-(GT)n introns: 5′ ss and 3′ ss are brought closer by base-pairing of AC and GT repeats. This intronic bridging can override the requirement of U2AF2 for 3′ ss recognition. (Right) Human (GGG)m-(CCC)n introns: 16% of human introns contain multiple copies of complementary G and C triplets near 5′ ss and 3′ ss, respectively, that stabilize the intronic structure. They may bridge the splice sites and facilitate splicing as the zebrafish (AC)m-(GT)n introns.
This arrangement of pairing across introns is broadly consistent with other examples of secondary structures that increase a particular splice site pairing (Goguel and Rosbash 1993; Libri et al. 1995; Charpentier and Rosbash 1996; Howe and Ares 1997; Chen and Stephan 2003; Graveley 2005). It is interesting to note that the pairing of complementary regions across exons has been associated with nonfunctional circular splicing in humans (Jeck et al. 2013). In zebrafish, this inhibitory cross-exon arrangement of AC and GT repeats is largely avoided (Fig. 2). Here, we propose that this pair of simple dinucleotide repeats is being utilized as paired elements that form a secondary structure across introns in the chordate phylogeny. While we concluded that this structure promoted constitutive splicing, we discovered several loci with architecture reminiscent of the Dscam1 system—upstream AC repeat followed by GT repeats in successive downstream introns (Graveley 2005). However, in none of the loci tested were we able to detect (AC)m-(GT)n-driven Dscam1-like mutual exclusion in zebrafish (data not shown).
We report striking differences in folding energies of introns that contain AC and GT repeats. Shuffling experiments strongly suggest that this stability is not just a consequence of composition, but rather the repeats have evolved to adopt arrangements that favor the formation of strong secondary structures. Although there have been reports of predicted secondary structure sequestering elements or reducing the distance between particular elements, at the time of this writing we are not aware of any reported cases of secondary structure bridging nuclear introns in vertebrates (Warf and Berglund 2010; Lovci et al. 2013). We discovered these signals in the introns of teleosts (ray-finned fish) but not in tetrapods (Fig. 6). The fact that AC and GT repeats co-occur frequently in Agnathans (lamprey) suggests an ancient origin of this mechanism (i.e., the last common ancestor of lampreys and fish precedes the tetrapod/teleost divergence). Self-splicing group II introns which are presumed to share ancient origins with nuclear introns also rely upon intronic structural elements to align 3′ ss or 5′ ss (Lambowitz and Zimmerly 2004). While the mechanism of (AC)m-(GT)n introns is found across the fish and lamprey genomes, individual instances of (AC)m-(GT)n introns are not highly conserved in the orthologous introns of related fish species. This lack of conservation observed between orthologous introns is likely due to the high background mutation rate associated with simple repeats. The expansions and contractions of AC repeats occur at a frequency of 1.5 × 10−4, about 10,000 times higher than other types of mutations (Shimoda et al. 1999; Conrad et al. 2011). In fish, this dimer is no more prone to expansion than in mammalian lineages; however, the degree of gain/loss is greater in fish (Weber and Wong 1993; Crawford and Cuthbertson 1996; Shimoda et al. 1999). The gain or loss of multiple repeats is rare in mammals, but in fish, 29% of all indel events involve five or more repeats. Given this bias toward creating short blocks of AC or GT repeats, it is possible to see how a hairpin that bridges introns could arise or be destroyed by a single mutational event. In the human orthologs of (AC)m-(GT)n introns, where the bridging structure was presumably lost, we see extended PPT and a higher frequency of purine-rich ESE-like sequences (Supplemental Fig. 9). U2AF2 is an activator that is localized to the 3′ ss through its binding to the PPT or, indirectly, through protein-protein interactions with SR proteins that bind ESEs (Singh et al. 1995; Zuo and Maniatis 1996). We confirm in vitro and in vivo that U2AF2 is essential for processing of the control intron but is, surprisingly, not required for the splicing of (AC)m-(GT)n structured introns (Fig. 5). The extended pyrimidine tracts and ESE motifs seen in the human orthologs of (AC)m-(GT)n introns suggests that they are more dependent on U2AF2 after the presumed loss of the (AC)m-(GT)n bridging structure that pairs splice sites across the introns (Supplemental Fig. 9).
Although this report is mostly focused on structure, it is likely that the effect of these RNA elements and proteins cannot be completely separated. The mutually exclusive splicing in the Drosophila Dscam1 depends on structure but is also regulated by proteins (Celotto and Graveley 2004; Olson et al. 2007). The hnRNP proteins, like most splicing factors, seem to recognize single-stranded RNA (Schenkel et al. 1988; Kumar and Wilson 1990; Trauger et al. 1990; Munroe and Dong 1992; Auweter et al. 2006; Huang et al. 2008). Yet, there are numerous splicing factors such as SR proteins, hnRNP proteins, and the factor YB-1 that assist RNA annealing events and even catalyze the rearrangement of a suboptimal structure into the optimal low-energy structures (Kumar and Wilson 1990; Munroe and Dong 1992; Lee et al. 1993; Skabkin et al. 2001; Shen and Green 2006). We have noted that (AC)m-(GT)n repeats are prevalent in teleosts and lampreys, but tetrapods have retained a slight enrichment of GT repeats in the 3′ ss region (Fig. 6). It has been described that GT repeats [(GT)16GGGCAG-3′ ss] of human apolipoprotein A-II could positively direct the splicing (Shelley and Baralle 1987). Our work suggests differences in the role of U2AF2 between lineages. It is possible that protein factors recognizing these GT repeats have supplanted the role of structure in the processing of these introns. CELF1 (also known as CUGBP), MBNL1, and TARDBP have all been implicated in binding GT-rich sequences (Marquis et al. 2006; Warf and Berglund 2007; Bhardwaj et al. 2013). Specifically, TARDBP interacts with GU-repeats at the 3′ end of cftr (cystic fibrosis transmembrane conductance regulator) intron 8 and promotes exon 9 skipping (Buratti et al. 2004; Groman et al. 2004); however, effects of GU-repeats at the 5′ end of introns are dependent on splice site strength and the presence of additional regulatory sequences (Passoni et al. 2012). Imbalance of GU-binding proteins results in splicing defect and diseases (Lee and Cooper 2009). As trans-acting factors like these become better characterized, it is likely that a greater role for repetitive sequences as cis regulatory elements in gene expression will be uncovered. Repetitive sequences comprise over two-thirds of the human genome, yet this class of sequences is underrepresented in genome assemblies and is often discarded in high-throughput analysis of gene expression (de Koning et al. 2011). We have presented a positive functional role for these repeats that precedes the divergence of fish from tetrapods. To our knowledge, this ancient class of introns is the first example of unrelated spliceosomal introns characterized by a significant structural motif, and this discovery helps connect the spliceosome and its substrates to structured RNA ribozymes that catalyze their own removal from transcripts.
Methods
Constructing and comparing position distributions
The computational techniques used here to construct intron/exon databases and positional distribution for hexamers were adapted from previous work (Lim et al. 2011). For every species, each entry in the Ensembl database consists of at most 600 nt: two 200-nt intronic flanks and two 100-nt exonic flanks on each side of the splice sites. In the case where intronic or exonic length is <400 or 200 nt, respectively, the sequence is divided by half and each half is assigned to its nearest splice site. All duplicated entries were screened and removed from the database. To allow fair comparison between human and a second species, only orthologous pairs of entries between the two species are considered. We used the UCSC Genome Browser tool LiftOver (Hinrichs et al. 2006), which parses the outputs of the multiple alignment tool BLASTZ and returns orthologous coordinates. Since the ultimate goal is to compare the shapes of each k-mer's distribution from two species around annotated splice sites, each numerical vector was standardized using z-score normalization. Exon and intron regions were normalized separately to eliminate k-mers’ enrichment in exons due to their protein coding function. The normalized vector was then compared by L1 distance. The L1 distance is calculated as the sum of the absolute value of the difference between two hexamers. In other words, higher L1 distance infers a greater difference of a hexamer between two species, thereby suggesting a potential change in its functional role across species. This process was repeated for AC and GT repeat hexamers on additional genomes listed in Figure 6. Introns were classified as (AC)m-(GT)n introns by the more stringent demand of ACACAC/GTGTGT within a window of 50 nt from the 5′ ss/3′ ss. Ensembl and UCSC Known Genes genome annotations were used to calculate average lengths of (AC)m-(GT)n introns across listed fish species (Supplemental Tables 1, 2). t-tests compared the average intron lengths in species where (AC)m-(GT)n were longer than average against the average intron lengths where introns were shorter than average. Zebrafish alternative splicing was characterized using publicly available RNA-seq from adult head (GSM977959) and tail (GSM977960) tissues. Read alignments and alternative splicing transcripts were generated using the TopHat-Cufflinks pipeline (Trapnell et al. 2012). Significant differential splicing events were determined using a χ2 test (P < 0.05) of read counts over splice junctions.
Co-enrichment
Co-enrichment of AC and GT repeats were calculated by comparing observed co-occurrences to expected co-occurrences predicted assuming (1) independence between AC and GT distribution in introns, and (2) uniform probability of occurrence of AC and GT repeats equal to genome wide average. Several repeat lengths (i.e., numbers of AC or GT repeats) were counted in zebrafish intronic windows of 200 nt flanking the splice sites, and co-enrichment was compared across the intron (Fig. 2, left panel) and exon (right panel).
Structure prediction
Introns were excised at the 5′ and 3′ ends and binned according to intron length. RNAfold (Zuker and Stiegler 1981), an RNA structure prediction program, was used to both generate predicted minimum free-energy structures for display and report the predicted ΔG of folding. The RNAfold program was used with default settings.
To test the contribution of the pairing of (AC)m-(GT)n hexamers in zebrafish or (GGG)m-(CCC)n triplets in humans toward the overall intronic hairpin structure, zebrafish introns with at least one ACACAC/GTGTGT hexamer pair and human introns with at least three GGG/CCC triplet pairs in the first and last 40 nt were each shuffled 1000 times. These shuffles maintained the same overall concentration of repeats (AC/GT in zebrafish and GGG/CCC in humans) and the overall nucleotide composition within each intron but were otherwise shuffled randomly using a custom Perl script. Each of the 1000 shuffled sets was folded with RNAfold, and the average ΔG of the predicted minimum free-energy structure was extracted to calculate a P-value.
RNase mapping
RNase mapping was performed as described (Brown and Bevilacqua 2005). Briefly, the RNA probes (172 nt of AC-GT or 201 nt of CON-GT and CON-pair) were transcribed from PCR products using a MEGAshortscript T7 kit (Ambion). To remove the 5′ phosphate, each RNA probe was treated with 10 units of calf intestinal alkaline phosphatase (New England BioLabs [NEB]) in dephosphorylation buffer (50 mM Tris-HCl pH 8, 10 mM MgCl2, and 0.1 M NaCl, supplemented with 20 units RNasin ribonuclease inhibitor [Promega]) at 37°C for 1 h. The RNA probes were then phenol/chloroform-extracted and gel-purified. To end-label the RNA probe, 20 pmol of each probe were incubated with 20 pmol ATP-[γ-32P] (3000Ci/mmol, 10 mCi/mL; PerkinElmer) and 10 units T4 polynucleotide kinase (NEB) in PNK buffer (70 mM Tris-HCl pH 7.6, 10 mM MgCl2, and 5 mM DTT, supplemented with 20 units RNasin) at 37°C for 1 h. The labeled RNA was subject to NucAway Spin Column (Ambion) cleanup, phenol/chloroform extraction, and gel purification. RNA probes were heated at 94°C for 3 min and cooled down on the bench top for at least 5 min to facilitate the structure formation. For a 10 μL enzymatic reaction, 2 × 105 cpm probe, 10 μg yeast RNA, RNA structure buffer (Invitrogen), and 3.3 units or 0.33 units RNase T1 (Ambion), or 0.2 or 0.02 μg RNase A (Ambion), or 0.1 units or 0.01 units RNase V1 (Invitrogen) were mixed, and incubated at room temperature for 5 or 15 min. To generate a hydrolysis ladder, probes were mixed with 10 μg yeast RNA and alkaline hydrolysis buffer (Invitrogen) and heated at 94°C for 5 min. The digested products were resolved in an 8.5% urea polyacrylamide gel, exposed to phosphorimager plates, and analyzed by Typhoon scanner (GE Healthcare).
Minigene reporter experiments
A three-exon, two-intron backbone, pZW4 (Wang et al. 2004), was used to generate splicing minigenes. The reporter constructs were made by inserting intron 5 of cep97 (with portions of exonic flanking sequence) or intron 16 of sacm1lb (with portions of exonic flanking sequence) into the middle exon of pZW4, creating a four-exon, three-intron construct (Fig. 4A,B), or replacing both introns and the middle exon of pZW4, creating a two-exon, single-intron construct (Fig. 4A,D). Transfections were performed by cationic lipid (Lipofectamine 2000; Invitrogen) into human embryonic kidney 293 cells. The RT-PCR was performed on total RNA using primers in the first and last exon to determine the degree of splicing for each construct. Intronic primers designed to extend in opposing directions were used to copy through the BP of the lariat. PCR products were run on agarose gels and stained by ethidium bromide. Cryptic products were identified and BP usage was mapped by sequencing the resulting PCR products. Reverse field images were analyzed by a UVP bioimaging system (UVP, LLC) or Image J (National Institutes of Health) to determine band intensities.
In vitro splicing
HeLa nuclear extract was prepared using publicly available protocols (Folco et al. 2012; and http://labs.umassmed.edu/moorelab). The zebrafish AC-GT splicing substrate was PCR-amplified by primers T7-a-F: CGAAATTAATACGACTCACTATAGGGAGACCCAAACGGAAACATCATCACCAC, and Rev-ab: CCTTCCTTCTGTGTGACGGCCACTCC; AdML81 (Bennett et al. 1992) was cloned into the pcDNA3 plasmid and linearized by Xba1. Both were then in vitro-transcribed by T7 RNA polymerase (Agilent Technologies) supplemented with UTP-[α-32P] (PerkinElmer). A 10-µL splicing reaction was 10 nM RNA substrate, 1 µL of 10× splicing buffer (20 mM MgOAc, 200 or 800 mM KOAc, 10 mM ATP, and 50 mM creatine phosphate), 4 µL nuclear extract with or without antibody as indicated. To block splicing with antibodies, we followed a protocol demonstrated to eliminate protein binding to a radiolabeled probe through pre-incubation with blocking antibodies (Lim et al. 2011). Splicing reactions were prepared in the absence of the RNA substrate with 200 or 600 ng of blocking MC3 monoclonal antibody against U2AF2 (also known as U2AF65; Santa Cruz Biotechnology), 600 ng antibodies against SF3B3 (Proteintech), or 600 ng normal rabbit IgG (Santa Cruz Biotechnologies) as a control, incubated at 30°C for 10 min before supplementing the substrate, and allowing splicing at 30°C for 60 min. The splicing reaction was terminated by adding 120 µL of splicing dilution buffer (100 mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, 1% SDS, 150 mM NaCl, and 300 mM NaOAc pH 5.2) and phenol/chloroform extraction. Following ethanol precipitation, the spliced RNA was analyzed by an 8% urea polyacrylamide gel and visualized with phosphorimager plates.
Polypyrimidine tract identification and scoring
PPTs were identified within 40-nt windows immediately upstream of the annotated 3′ ss. The algorithm used for identifying and scoring the PPTs was described in a previous study (Clark and Thanaraj 2002). Significance between PPT score distributions was determined using the Wilcoxon test.
In vivo knockdown by morpholino oligo
Freshly spawned wild-type zebrafish embryos were collected and stored in egg water (1.2 g Instant Ocean Aquarium Sea Salt Mixture, 500 μL methylene blue in 20 L ddH2O) for injection. The morpholino oligo sequence used in this study is AACTCGTCAAAGTCTGACATTTTCC for u2af2a and TCATCAAAATCAGACATCCTGGTGT for u2af2b. 0.5–1 nL of 0.8 mM morpholino oligos, mixed with phenol red for visualization, were injected into the yolk before the four-cell stage. Embryos were then incubated at 28°C for 24 or 48 h prior to RNA extraction for analysis.
RNA-seq differential expression workflow
Five μg of total RNA from the 24-h control or morpholino-injected whole zebrafish embyros were treated with the RiboMinus Eukaryote kit, v2 (Life Technologies) to remove the ribosomal RNAs. The resulting 620 ng RNA was then treated with 0.5 units/μg RNase III (Ambion) at 37°C for 15 min to homogenize the RNA. The reaction was terminated by phenol/chloroform extraction followed by ethanol precipitation. To build a sequenceable library, the resuspended RNA was (1) reverse-transcribed into cDNA by SuperScript II (Invitrogen) and random 9-mers, (2) made into double-stranded cDNA with the NEBNext mRNA Second Strand Synthesis Module (NEB), (3) made into blunt-end dsDNA with the NEBNext End Repair Module (NEB), (4) made into dsDNA with a single A overhang with NEBNext dA-Tailing Module (NEB), (5) ligated with Illumina sequencing adaptors with NEBNext Quick Ligation Module (NEB), and (6) amplified to meet the sequencing requirement by NEBNext High-Fidelity 2× PCR Master Mix (NEB) and two outer primers recognizing the ligated Illumina adaptors (5′-AATGATACGGCGACCACCGAGATCTACAC and 5′-CAAGCAGAAGACGGCATACGAGAT). Reactions were cleaned up using Agencourt AMPure XP beads (Beckman Coulter) between steps. The library was sent to Axeq Technologies (Macrogen) for HiSeq 100-bp paired-end sequencing.
We used the following procedures to compute normalized counts and calculate differences in expression values for genes in RNA-seq samples.
-
Download zebrafish genome Zv9/danRer7 and a GTF annotation from the UCSC Genome Browser (Mangan et al. 2008) (table Ensembl Genes).
-
Reads from each sample were aligned to the genome using STAR aligner (Dobin et al. 2013) using default settings with the exception of: outFilterMismatchNoverLmax, which was set to 0.05; and outFilterMultimapNmax, which was set to 1.
-
To count fragments that overlap a particular transcript in the GTF file, featureCounts (Liao et al. 2014) software was run with default settings with the exception of the following parameters: isPairedEnd = TRUE; minMQS = 10.
-
Standard DESeq2 (Love et al. 2014) pipeline was applied to the counts.
Data access
The RNA-seq data sets generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP060685.
Acknowledgments
We thank Dr. Heather Thompson, Dr. Rebeka Merson, and members of the Fairbrother lab for helpful comments. The lab is supported by National Institutes of Health (NIH) grants R01GM095612, R01GM105681, and by Brown University through the use of the OSCAR cluster and the genomics core facility (8P30GM103410).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.181008.114.
- Received July 5, 2014.
- Accepted November 10, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








