
The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing
- Marcin Cieslik1,
- Rashmi Chugh2,
- Yi-Mi Wu1,
- Ming Wu1,3,
- Christine Brennan1,
- Robert Lonigro1,
- Fengyun Su1,
- Rui Wang1,
- Javed Siddiqui1,
- Rohit Mehra1,
- Xuhong Cao1,3,
- David Lucas4,
- Arul M. Chinnaiyan1,3,4,5,6,7 and
- Dan Robinson1,7
- 1Michigan Center for Translational Pathology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 2Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA;
- 3Howard Hughes Medical Institute, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA;
- 4Department of Pathology, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA;
- 5Comprehensive Cancer Center, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA;
- 6Department of Urology, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA
- Corresponding author: arul{at}med.umich.edu
Abstract
RNA-seq by poly(A) selection is currently the most common protocol for whole transcriptome sequencing as it provides a broad, detailed, and accurate view of the RNA landscape. Unfortunately, the utility of poly(A) libraries is greatly limited when the input RNA is degraded, which is the norm for research tissues and clinical samples, especially when specimens are formalin-fixed. To facilitate the use of RNA sequencing beyond cell lines and in the clinical setting, we developed an exome-capture transcriptome protocol with greatly improved performance on degraded RNA. Capture transcriptome libraries enable measuring absolute and differential gene expression, calling genetic variants, and detecting gene fusions. Through validation against gold-standard poly(A) and Ribo-Zero libraries from intact RNA, we show that capture RNA-seq provides accurate and unbiased estimates of RNA abundance, uniform transcript coverage, and broad dynamic range. Unlike poly(A) selection and Ribo-Zero depletion, capture libraries retain these qualities regardless of RNA quality and provide excellent data from clinical specimens including formalin-fixed paraffin-embedded (FFPE) blocks. Systematic improvements across key applications of RNA-seq are shown on a cohort of prostate cancer patients and a set of clinical FFPE samples. Further, we demonstrate the utility of capture RNA-seq libraries in a patient with a highly malignant solitary fibrous tumor (SFT) enrolled in our clinical sequencing program called MI-ONCOSEQ. Capture transcriptome profiling from FFPE revealed two oncogenic fusions: the pathognomonic NAB2-STAT6 inversion and a therapeutically actionable BRAF fusion, which may drive this specific cancer's aggressive phenotype.
Despite advances in tissue preservation and handling, it remains a challenge to obtain RNA of sufficient integrity from clinical specimens (Medeiros et al. 2007; Turashvili et al. 2012). Oncological tissues procured via needle core biopsies and preserved as formalin-fixed paraffin-embedded (FFPE) blocks remain problematic for the most commonly used RNA-seq protocols (Lister et al. 2008; Mortazavi et al. 2008; Nagalakshmi et al. 2008), which contrasts with their routine use in cell lines. Due to the utility of expression profiles in the diagnosis, prognosis, and therapy of cancer, there is a growing clinical need for methods that produce reliable data from samples that vary in source material and quality (Bittner et al. 2000; Armstrong et al. 2002). To date, no protocol has been shown to robustly and accurately measure absolute gene expression from degraded RNA, which has impeded the use of RNA-seq to profile the expression of clinical samples. As neither mRNA enrichment “poly(A)” nor rRNA depletion “Ribo-Zero” (Zhang et al. 2012) libraries can be reliably generated from degraded and cross-linked RNA, novel protocols are needed to unlock these valuable data for precision medicine approaches or retrospective studies.
An alternative approach is to directly select for known transcripts using complementary capture probes. Direct target enrichment protocols were initially designed to capture the exome from the total genomic DNA for the purpose of cost-effective clinical resequencing (Choi et al. 2009) and were next adapted for cDNA targets (Ravo et al. 2008; Ueno et al. 2012). In capture sequencing, each transcript of interest is targeted with an excess of probes at multiple positions, which makes transcript recovery possible even if the poly(A) tail was lost. Recently, targeted RNA sequencing was suggested as a method to comprehensively sample low-abundance isoforms (Mercer et al. 2012; Halvardson et al. 2013; Fu et al. 2014) and even measure gene expression (Cabanski et al. 2014). However, the recommendation of a novel transcriptome profiling protocol for routine use in a clinical or research setting requires careful examination of its relative merits on a wide range of metrics (Mullins et al. 2007; Zeng and Mortazavi 2012; Adiconis et al. 2013; Zhao et al. 2014). It is critical that the recommended method is largely compatible with poly(A) RNA-seq and Ribo-Zero libraries as these are most commonly used for research and by The Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Research Network 2008).
Results
We developed the exome-capture (short “capture”) RNA-seq library preparation protocol as a modification to our clinical poly(A) selection (short “poly(A)”) RNA-seq procedure (Fig. 1A). The protocols share a number of steps but differ at two important stages (Methods). Briefly, for poly(A) selection, oligo(dT) beads are used at the beginning of the workflow to enrich for spliced and polyadenylated mRNAs. This step is omitted for capture transcriptomes; for which, alternatively, enrichment is done after the main enzymatic steps of library construction. Unique to capture transcriptomes is an overnight capture reaction (RNA-DNA hybridization) using exon-targeting RNA probes, followed by a washing step, and an additional set of PCR cycles. After the final PCR reaction, both types of libraries are ready for clustering on an Illumina flow-cell (Fig. 1A).
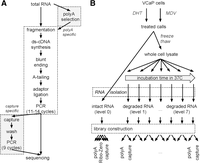
The exome-capture transcriptome protocol. (A) Flow-chart of library preparation protocols. Steps unique to each protocol are highlighted. Enrichment for mRNA occurs at the RNA or cDNA stage, respectively, for poly(A) and capture RNA-seq. (B) Controlled in vitro degradation through cell lysis and warm incubation. VCaP cells were treated with DHT or MDV3100. Intact RNA, RNA integrity number (RIN) 10, was extracted, and libraries were prepared in technical triplicates. In parallel, RNA was degraded by warm incubation for increasing amounts of time. Paired poly(A) and capture libraries were prepared from the same RNA at each degradation level.
Concordance of capture and poly(A) transcriptomes from intact RNA
To assess the similarity, consistency, and efficiency of transcriptomes obtained using the exome-capture and poly(A)-selection protocols, we prepared a total of 12 libraries (technical triplicates) from perfectly intact RNA (RIN 10.0, RNA degradation level 0) (Fig. 1B; Supplemental Fig. S1). Total RNA was extracted from VCaP prostate cancer cells treated with dihydrotestosterone (DHT) or enzalutamide (MDV3100, short MDV). First, we looked at alignment rates and the degree of strand-specificity (Parkhomchuk et al. 2009; Levin et al. 2010), or “strandedness.” We found that libraries from both protocols have high and reproducible alignment rates (∼85%) and almost perfect strandedness (Fig. 2A). Next, we computed genomic distributions of the aligned fragments to gauge the on-target performance of the protocols. For both protocols, at least 95% of all aligned fragments were shown to overlap known exons (Fig. 2B, left). Since the reference genome does not include rRNA coding loci, we conclude that the protocols are equivalent in providing approximately the same number of useful reads for a given depth of sequencing.
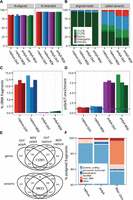
Similarity of poly(A) and capture transcriptomes from intact RNA. Properties of fragments from both types of libraries. Separate bars (colors) for each replicate in A,C,D. (A) Alignment rates and library strand-specificity (% fragments aligned to the transcribed strand). (B) Types of genomic alignment regions by fraction of assigned fragments and fraction of discovered variants. (C) Efficiency of rRNA depletion (% fragments aligning to ribosomal RNA). (D) Overrepresentation of poly(A) and poly(T) hexamers. (E) Global concordance of detected genes and called variants within all exonic regions. (F) Fraction of assigned reads by biological gene category.
We estimated the amount of rRNA by aligning reads to the rRNA precursor sequences (Methods). Positive selection for poly(A) is an efficient method for the removal of rRNA sequences as these are never polyadenylated. As expected, poly(A) libraries were found to be virtually free of rRNA (<1%), while capture libraries were found to contain ∼10% rRNA (Fig. 2C), which makes them superior to those from alternative RNA-seq protocols, e.g., DSN (40%) and Ribo-Zero (23%) in Langevin et al. (2013) (Supplemental Fig. S2). In contrast, enrichment using oligo(dT) introduces a strong and reproducible 7.5-fold overrepresentation of adenine homopolymers (Fig. 2D). In total, the genomic origin can be determined for 95% of reads from capture compared to <90% from poly(A) libraries.
Next, we computed the overlap between poly(A) and capture libraries for detectably expressed genes and single nucleotide variants (SNV) within all exons (Fig. 2E; Methods). The majority of genes, 80% (over 11,000 genes), can be detected in both, 15% (2452 genes) are unique to poly(A), and 4.5% (688 genes) are unique to capture libraries. Conversely, of the total 14,271 variants, 6% (857) were unique to poly(A) and 12% (1646) to capture. Protein coding genes are currently the most clinically relevant and actionable gene “biotype” (Harrow et al. 2012), while noncoding RNAs are emerging as robust biomarkers. To test whether these are adequate, we tallied aligned reads in capture, poly(A), and Ribo-Zero libraries by gene biotype (Fig. 2F). We observed no differences in the proportion of reads originating from protein coding genes between poly(A) and capture. Compared to poly(A), capture libraries contained more reads from long noncoding RNAs.
In summary, capture and poly(A) transcriptomes from identical and intact RNA are similar. They cover similar genomic regions and detect overlapping sets of SNVs. Differences in coverage are observed within introns, intergenic regions, and for non-protein-coding genes. As expected, noncoding genes are mostly nonpolyadenylated and are enriched in Ribo-Zero libraries (Cui et al. 2010). Importantly, capture transcriptomes are more sensitive in calling variants within exons (Fig. 2B, right; Fig. 2E).
Quantitative gene expression profiles from exome-capture transcriptomes
Next, we sought to establish whether capture transcriptomes provide precise estimates of absolute gene expression. We tested whether quantification was limited to genes included in the probe design (Methods) and found that genes that are captured are detected at the same rate in poly(A) and capture libraries (∼11,000 in VCaP), whereas genes (∼2000 in VCaP) that are not captured are largely missing. Hence, we decided to focus all subsequent quantitative analyses on the captured genes.
First, we benchmarked the protocols in terms of technical reproducibility and found that both show excellent agreement (Supplemental Figs. S3, S4). We also plotted mean-variance trends (Supplemental Figs. S5, S6) and expression-level histograms (Supplemental Fig. S7) and found, respectively, no significant differences in variability and dynamic range between the two library types. We compared the distributions of gene expression levels from capture, poly(A), and Ribo-Zero libraries using Q-Q plots and observed only small deviations for the most highly expressed genes (Fig. 3A).
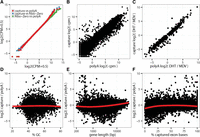
Agreement of absolute and differential gene expression. Expression levels were quantified by counting the number of aligned fragments within captured exonic regions and converted to the log2 of counts per million (log2[cpm]). Treatment log2 fold-changes were estimated through linear modeling. (A) Pairwise Q-Q plots comparing the distributions of gene expression levels. (B) Agreement of absolute levels of transcript abundance log2(cpm). (C) Agreement of differential gene expression between DHT-treated and ablated cells (MDV treatment) (log2 fold-changes). (D–F) Observed differences between capture and poly(A) expression estimates are not driven by GC content, gene length, or fraction of exon bases with target probes.
Finally, we directly compared expression estimates between capture and poly(A) libraries (Fig. 3B) and found them in very good agreement across the full dynamic range of gene expression levels. We find that expression estimates from both library types are within a factor of two for the majority of genes (>87%). Genes with higher expression levels in the capture libraries were identified as histones and small nucleolar RNAs (Fig. 3B; Supplemental Table S1), which was reported for Ribo-Zero and DSN libraries (Miller et al. 2013; Zhao et al. 2014) and is explained by their unique biology: Histone mRNAs in metazoans are not polyadenylated (Yang et al. 2011), while polyadenylation of snoRNAs is a signal for their degradation (LaCava et al. 2005). A small number of genes were found to be underestimated in capture libraries. The majority of those were inadequately captured (see below).
Next, we focused on quantitative estimates of differential gene expression. To identify androgen receptor (AR)-regulated genes, we estimated log2 fold-changes (Smyth 2005; Law et al. 2014) between dihydrotestosterone- and enzalutamide-treated cells (Methods). Both the estimated log2 fold-changes (Fig. 3C) and differential expression P-values (Supplemental Fig. S8) are in excellent agreement between capture and poly(A) libraries. The log2 fold-changes estimated from both protocols match closely across 10 orders of magnitude (Fig. 3C), while P-values of differential expression are precise even for genes with small effect sizes (Supplemental Fig. S8). Known AR targets were found among the most up- and down-regulated genes including TMPRSS2 (>10-fold up) and MYC (greater than twofold down) (Supplemental Table S2). We see no evidence of saturation. For example, the gene PGC is estimated to be induced 824-fold in the poly(A) libraries compared to 1096-fold in the capture libraries (Fig. 3C).
Recent studies have revealed complex biases inherent to RNA-seq. We decided to look into the most common sources of bias in RNA-seq libraries: GC content (Risso et al. 2011) and gene length (Oshlack and Wakefield 2009), and also a factor unique to capture libraries, the “capture ratio,” i.e., the percentage of targeted exonic bases. Our results indicate that capture efficiency is not significantly biased by GC content (Fig. 3D), gene length (Fig. 3E), or capture ratio (Fig. 3F). Only minor trends were revealed; a small proportion of genes with low capture ratio (<25%) were inadequately captured, resulting in the underestimation of their expression levels (Fig. 3F). Conversely, long genes are underestimated in poly(A) libraries (Sigurgeirsson et al. 2014), which can be attributed to a loss of 5′ transcript ends clearly present even in RIN 10 RNA (Fig. 3E). Altogether, these results suggest that exome-capture RNA-seq provides precise and largely unbiased estimates of gene expression for the majority of captured genes.
Improved performance of capture libraries from low-quality RNA samples
Our initial experience with capture RNA-seq libraries accrued through the clinical MI-ONCOSEQ project (Roychowdhury et al. 2011) indicated their improved performance for fusion calling and greater reliability in samples of low RNA quality. To test this rigorously, we performed controlled in vitro degradation of RNA (Fig. 1B) by preparing paired capture and poly(A) libraries at increasing levels of RNA degradation (RIN 2–9) (Fig. 1B; Supplemental Fig. S1; similar to Thompson et al. 2007; Opitz et al. 2010; Sigurgeirsson et al. 2014).
To detail how RNA degradation affected estimates of gene expression, we first compared degraded (level 7) capture, Ribo-Zero, and poly(A) libraries with reference poly(A) transcriptomes (level 0). We observed good agreement (R > 0.85) for the majority of pairwise comparisons (Fig. 4A; Supplemental Fig. S9). Importantly, the highest correlation with reference poly(A) was observed for degraded capture, not poly(A), transcriptomes (R = 0.96). Degraded samples correlated less well, which suggests that degradation is associated with significant technical variability. To assess this further, we quantified the average variability of gene expression (coefficient of variation, CV) across libraries from the same RNA but a range of degradation levels (Fig. 4B). As expected, we found that capture libraries were significantly more precise (less variable). We performed unsupervised clustering of DHT- and MDV-treated samples to assess whether technical variation from RNA degradation obscured biological differences (Fig. 4C). If technical variation is sufficiently low, samples will cluster by treatment “DHT/MDV” and not by RNA quality “(0–9).” This was the case for capture libraries, which partitioned by treatment first and RNA quality second. On the contrary, poly(A) libraries were inadequately controlled and clustered predominantly by RNA quality, obscuring the treatment (Fig. 4C). Next, we assessed the impact of RNA degradation on the sensitivity of calling SNVs. For each sample, we computed the fraction of SNVs that were successfully detected (Fig. 4D; Methods). We observed that the recall of variants rapidly declined with RNA degradation for poly(A) but not for capture libraries (Fig. 4D). We reasoned that the poor performance of poly(A) libraries is likely due to a decrease in coverage of the variant positions. To test this, we computed the fraction of unique fragments (Fig. 4E) and the distribution of fragments along the gene body (Fig. 4F). As expected, we found that library complexity was negatively correlated with RNA quality. The deterioration was relatively mild in capture libraries but very strong in poly(A) libraries, for which up to ∼90% of the fragments were duplicates (Fig. 4E). Uniformity of fragment distribution and coverage along the gene body is critical for identifying full-length transcripts and calling variants at the 5′ or 3′ transcript ends. We found that intact poly(A) samples have a 3′ bias (Fig. 4F), which, as reported previously (Popova et al. 2008; Opitz et al. 2010; Sigurgeirsson et al. 2014), is associated with degradation. Conversely, capture libraries have higher coverage at the 5′ end, and gene coverage is robust to RNA degradation. This bias is “by design” since hybridization probes are placed infrequently within long 3′ UTR regions (Supplemental Fig. S10).
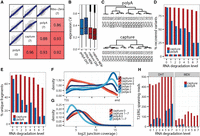
Improved performance of exome-capture transcriptomes from low quality RNA samples. (A) Correlation of absolute levels of gene expression (log2[cpm]) between a reference library from intact RNA (poly[A] level 0) and libraries from degraded RNA (level 7). (B) Impact of RNA degradation on gene expression accuracy measured as the average coefficient of variation (CV)—larger values indicate more variable measurements. (C) Impact of expression accuracy on the unsupervised clustering of samples with biological differences confounded by technical variation. (D) Sensitivity of detection of single nucleotide variants in libraries of varying RNA quality. (E) Library complexity estimated as the percentage of unique (nonduplicate) fragments among all counted fragments. (F,G) Assessments of uniformity of transcript coverage. (F) Smooth density estimate of read start positions along the scaled gene bodies (genes <10 kb were excluded). (G) Distribution of splice junctions by depth of coverage. (H) Sensitivity of detecting the TMPRSS2-ERG fusion (junction coverage).
Finally, we compared the two library types for their ability to detect splice junctions and gene fusions. We quantified the coverage of known splice junctions as a proxy for their likelihood of being de novo discovered (Fig. 4G). We found that the coverage of splice junctions was highest in capture libraries independent of RNA quality. Next, we assessed whether the differences in splice junction coverage actually influenced our ability to detect TMPRSS2-ERG, a known gene fusion (Tomlins et al. 2005). In DHT-treated samples, TMPRSS2-ERG is induced and expressed at a very high level. Correspondingly, the fusion was detected regardless of RNA quality and library type (Fig. 4H, left). In MDV (bicalutamide)-treated samples, where TMPRSS2-ERG is repressed, the fusion was reliably detected in all capture libraries, but only two out of seven degraded poly(A) libraries (Fig. 4H, right).
Application of capture transcriptomes in a rapid autopsy prostate cancer cohort
To confirm the in vitro results in a clinical setting, we extended the evaluation of capture RNA-seq to flash frozen tissue and FFPE blocks from an autopsy cohort of 13 prostate cancer patients. We sequenced a total of 29 samples divided into three types of libraries: capture FFPE, capture frozen, and poly(A) frozen (Supplemental Table S3). Chiefly, we wanted to establish if capture FFPE allowed for precise estimates of gene expression and whether the capture protocol provided a substantial improvement over poly(A) in frozen samples.
To begin with, we probed if the patient libraries were sufficiently depleted of ribosomal RNA (Fig. 5A). Levels of rRNA were found to be variable (2%–24%) in capture libraries and, as expected, very low (∼1%) in poly(A) libraries. As previously reported for Ribo-Zero (Zhao et al. 2014), we observed that capture libraries from FFPE contained significantly less rRNAs compared to libraries from frozen tissue. The lower rRNA content was also reflected in higher alignment rates (Fig. 5B). We developed a compound measure of library quality, which we term “fragment diversity.” This normalized score is sensitive to library complexity, 3′ bias, coverage, and insert size (Methods). We found capture libraries to be more diverse (P-value < 1 × 10−16) than poly(A) in frozen tissue (Fig. 5C). As anticipated, FFPE libraries were of lowest quality due to their limited size distribution (Supplemental Fig. S11) and complexity (Supplemental Fig. S12).
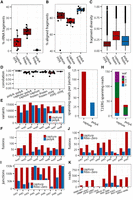
Assessment of capture transcriptomes from clinical frozen and FFPE samples. (A–C) Comparative analysis of paired capture and poly(A) libraries (grouped by patient) derived from FFPE blocks and frozen tissue: (A) efficiency of rRNA depletion; (B) alignment rates; (C) fragment diversity (FD)—a compound measure of transcriptome quality sensitive to coverage, complexity, and insert size; more complex and well-covered libraries have higher FD values. (D) Within patient correlation of gene expression (log2[cpm]) by library type (poly(A) vs. capture) and source material (frozen vs. FFPE). (E,F) Sensitivity of libraries for detecting genetic changes by patient from frozen libraries: (E) number of called variants; (F) number of called candidate fusions. (G,H) Robustness of fusion detection: (G) average read support per fusion; (H) number of supporting reads for each cohort patient with the TMPRSS2-ERG fusion detected. (I,J) Paired capture and Ribo-Zero libraries from FFPE: (I) number of detected splice junctions; (J) number of called candidate fusions. (K) Selected candidate oncogenic fusion for each patient (read support).
We next assessed the consistency of gene expression between matched libraries from the same patient. For one patient, all three library types were made, including duplicates of capture FFPE. Technical reproducibility of capture FFPE libraries was very high (R = 0.95), as was the correlation between capture FFPE and capture frozen (R = 0.93). The capture frozen library was in excellent agreement with the poly(A) frozen library (R = 0.93), despite systematic differences in read distribution (Supplemental Fig. S13). Next, we compared matched libraries from all cohort patients. We observed high correlations (R > 0.80) for all but two comparisons (Fig. 5D) and only small differences in variability between the types of compared libraries. The correlation of capture and poly(A) libraries was as good as the correlations between frozen libraries (Fig. 5D). Libraries from FFPE were also highly correlated with libraries from frozen tissue, irrespective of frozen library type. In summary, useful gene expression data was obtained from all frozen and all but one FFPE samples.
Finally, we compared capture and poly(A) libraries in terms of their ability to call variants (SNVs) and detect fusions from frozen clinical samples. For each patient, we computed the number of variants (Fig. 5E) and fusions (Fig. 5F) found in either library type. Informing our in vitro cell line results (Fig. 4D,H), we found that SNV calling was more sensitive in capture libraries, with thousands of variants called reliably only in capture libraries. Similarly, we nominated significantly more candidate fusions in capture libraries. Since fusion junctions are likely artifacts if they are supported by a small number of fragments, we also calculated the average number of spanning reads per junction (Fig. 5G). We found that fusions in capture libraries had threefold higher read support. The ETS gene family fusions are detected in over 50% of prostate cancer patients (Tomlins et al. 2005). We detected one of the ETS fusions (Supplemental Fig. S14A) in four out of eight (50%) patients from frozen capture compared to three from poly(A) (Fig. 5H). Importantly, the junction read support was significantly higher for capture libraries (Fig. 5H). These data are consistent with our observations from anti-androgen-treated VCaP cells (Fig. 4H), considering that many prostate cancer patients receive anti-androgen treatment.
Capture transcriptomes for robust fusion discovery from FFPE
To determine the sensitivity of capture for the detection of fusions from FFPE, we prepared matched capture and Ribo-Zero libraries for nine patient samples with putative oncogenic fusions. First, we assessed transcriptome coverage and uniformity by comparing numbers of detected splice junctions (Fig. 5I). Coverage of Ribo-Zero libraries was poor on average and inconsistent across samples, whereas capture libraries showed excellent reproducibility. Next, we looked at the total number of putative fusions. For all patients, significantly more fusions were nominated in capture libraries (Fig. 5J). To assess the clinical implications, we identified a known or candidate oncogenic fusion for each patient and counted the number of reads spanning the chimeric junction (Fig. 5K). Critically, the oncogenic fusion was detected in only three of nine Ribo-Zero libraries and with low number read support. Robust detection of fusions is necessary if driver fusions are functional at a low level of expression, such as EML4-ALK (Soda et al. 2007) and BRAF (Tian et al. 2011) fusions. Previously, we have identified the NAB2-STAT6 fusion as the defining characteristic of solitary fibrous tumors (SFT) (Robinson et al. 2013). An unusually aggressive case of SFT was referred to us for clinical cancer sequencing as part of the MI-ONCOSEQ program (Roychowdhury et al. 2011). The soft tissue mass metastasized within 6 mo to the lungs (Fig. 6A), and pathology revealed that the tumor lacked characteristics of SFT, such as low mitotic rate, rich vascularization, and CD34+ spindle-shaped cells. Mimicking a small cell sarcoma, it comprised closely spaced sheets of uniform, small undifferentiated cells, which were interspersed by rich vascular stroma and large zones of necrosis (Fig. 6B). We readily detected the NAB2-STAT6 fusion expressed at a very high level (over 2000 spanning reads) (Fig. 6C; Supplemental Fig. S14B). A more detailed look at the transcriptome revealed an in-frame BBS9-BRAF fusion (Fig. 6D; Supplemental Fig. S14B) with an intact kinase domain and recurrent truncation of the Ras binding domain (Poulikakos et al. 2011). Together, it is plausible that the BBS9-BRAF fusion is activating, contributes to the malignancy, and may be sensitive to either sorafenib or MEK inhibitors (Palanisamy et al. 2010). In summary, capture RNA-seq allows for robust yet unbiased detection of fusions from clinical FFPE specimens including rare and lowly expressed fusions.
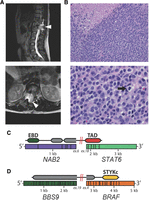
Clinically relevant gene fusions from FFPE in a case of solitary fibrous tumor. (A) MRI of the spine reveals a spinal canal mass with extradural extension from T10–T12 with mass effect and compression along the spinal cord (arrowhead). Recurrent disease caused cord compression at the T12–L1 right neural foramen. (B) The tumor mass comprises sheets of highly mitotic undifferentiated cells with rich vascular stroma and extensive zones of necrosis (upper left). High-power micrograph (bottom) illustrates the cytological features of pleomorphic small round cells with ill-defined eosinophilic cytoplasm, prominent nucleoli, and numerous mitotic figures (arrow). (C) NAB2-STAT6 is the defining oncogenic fusion in SFT. The trans-activating domain of STAT6 is highlighted in red, the EGR1 binding domain of NAB2 in green. (D) The BBS9-BRAF fusion is likely oncogenic as it retains the kinase domain of BRAF (yellow) and has a truncation of the Ras binding domain. BRAF fusions are typically expressed at a lower level, and this rearrangement was detected with 16 reads.
Discussion
Our data suggest that capture RNA-seq provides distinct advantages over poly(A) and Ribo-Zero RNA-seq in the clinical setting. A major difficulty in clinical RNA sequencing is the low quality of RNA isolated from clinical fresh frozen and FFPE samples. We and others have shown that even minimal RNA “nicking” has a profound negative effect on poly(A) libraries that is not addressed by the typical recommended threshold of RIN 8 (Zeng and Mortazavi 2012; Sigurgeirsson et al. 2014). Capture libraries are more robust to input RNA quality; splice-junctions, fusions, and variants can be comprehensively detected in the most degraded samples, while gene expression estimates remain precise and highly concordant with those from poly(A) and Ribo-Zero RNA-seq. We expect that the better success rate of capture RNA-seq will further the detection of expression signatures from frozen clinical samples and FFPE specimens. In agreement with previous studies in cell lines, we show on clinical specimens that target capture significantly improves the sensitivity of gene fusion detection (Ueno et al. 2012).
Limited amounts of starting material from clinical specimens represent a barrier to complex transcriptomes (Gertz et al. 2012; Ramsköld et al. 2012). We found that capture RNA-seq can accommodate the average RNA yields from five FFPE slides (0.5–40 µg), which is sufficient for good coverage and complexity. Even when frozen tissue is available, target capture outperforms poly(A) for SNV calling, thanks to its excellent coverage and complexity within coding regions (Zhao et al. 2014). Finally, capture RNA-seq opens up the possibility for clinical expression profiling of transcripts that are not predominantly polyadenylated, such as circular, enhancer, and long-noncoding RNAs.
Methods
Clinical samples
Samples were collected with informed consent and prior institutional review board approval. Prostate tissues were from the radical prostatectomy series and the Rapid Autopsy Program, which are both part of the University of Michigan Prostate Cancer Specialized Program of Research Excellence (SPORE) Tissue Core. The solitary fibrous tumor sample was obtained as archival tissue FFPE blocks. All CPRC specimens were obtained at rapid autopsy from men who died of lethal castrate resistant metastatic disease. Hematoxylin and eosin (H&E)-stained FFPE and frozen sections were reviewed to identify blocks with highest tumor content, a level was taken for H&E staining, and consecutive 3 × 10 µm sections were cut for RNA isolation. All H&E-stained levels were reviewed to confirm tumor/normal content before RNA isolation. RNA was isolated using the Qiagen RNeasy FFPE kit (cat. no. 73504).
Cell culture
The immortalized prostate cancer VCaP cell line was obtained from the American Type Culture Collection and was grown in DMEM (Invitrogen) and supplemented with 10% fetal bovine serum (FBS) with 1% penicillin-streptomycin. Before the treatments, cells were grown in androgen-depleted media lacking phenol red and supplemented with 10% charcoal-stripped serum and 1% penicillin-streptomycin. After 24 h, cells were treated either with androgen (1 nM 5α-dihydrotestosterone) or anti-androgen (enzalutamide). Cells were harvested for RNA isolation at 24 h post-treatment. RNA was isolated using the Qiagen AllPrep kit (cat. no. 80404).
RNA degradation
For controlled in vitro RNA degradation, after harvesting cells were frozen and thawed two times. The whole-cell lysate was incubated at 37°C for increasing amounts of time from 5 min to 6 h, after which RNA integrity was measured (Supplemental Fig. S1) and RNA was isolated as for the intact libraries.
Library preparation and sequencing
Details of the capture RNA-seq and poly(A) RNA-seq library preparation protocols are provided as Supplemental Material. Briefly, for capture libraries, we start with 0.1–3 µg of total RNA and proceed through first-strand synthesis, second-strand synthesis, end repair, A-tailing, adapter ligation, size selection on a 3% agarose gel, uridine digestion, hybridization to capture probes, washing, and a final PCR step. The stranded capture and poly(A) libraries were sequenced on an Illumina HiSeq 2500 using V3 chemistry.
Alignment
All the paired-end reads were aligned to the human reference GRCh37 augmented by splice junctions from Ensembl (Flicek et al. 2012) 75 using STAR 2.3 (Dobin et al. 2013) with default settings in two-pass alignment filtering mode “--outFilterType BySJout.” Only primary alignments were kept, duplicate fragments were marked using SAMtools (Li et al. 2009), and BAM files were sorted using novosort (http://www.novocraft.com/products/novosort/). The number of reads spanning each splice junction was obtained from the “SJ.out.tab” file provided by STAR.
Fragment quantification
All fragment quantifications were computed using featureCounts (Liao et al. 2013) (in stranded “-2,” paired-end, and “intersection_ nonempty” mode) (Anders et al. 2015). Briefly, in this method a fragment is assigned to a gene if it overlaps features of that gene only. Features are typically exons, and this definition was used for defining which genes are expressed (Figs. 2E, 3A). We also defined features as the genomic intersection between reduced/flattened exons (GenomicRanges) (Lawrence et al. 2013) and the captured regions. The latter definition was used for quantification of absolute and differential gene expression levels. Ensembl 75 annotations for exon type (“CDS” or “UTR”) and gene “biotype” (“protein_coding,” etc.) were used for all quantifications.
Strand-specificity (strandedness)
To estimate the strandedness of the library, the total number of assigned reads was counted for the correct “-s 2” and for flipped “-s 1” orientation, and strandedness was computed as: correct/(correct + flipped).
Ribosomal content
To estimate the fraction of ribosomal RNA (rRNA) fragments in each library, we aligned reads (using STAR) to a small reference including ribosomal sequences (NR_003286.2, NR_003287.2, NR_023379.1, NR_003285.2, NR_046235.1). The fraction of rRNA was estimated from the number of fragments aligning to the ribosomal reference from a random sample of 1 million fragments from each library.
Gene expression
Counts for each gene were transformed into cpm (counts per million) values using the “voom” (Law et al. 2014) function. To identify differentially expressed genes and calculate log2 fold-changes between the triplicate DHT- and MDV-treated libraries or capture and poly(A) libraries (see experimental design, Fig. 1B), we employed the standard limma (Smyth 2005) approach with cpm + precision values as input, with all default parameters. For calculations of dynamic range (Supplemental Fig. S7), fragments per kilobase per million (FPKM) were calculated using edgeR (Robinson et al. 2010) and gene lengths as reported by featureCounts.
Sub-exon path calculations
Tools necessary for the following computations were released as part of the sepath package (https://github.com/mcieslik-mctp/sepath/) implemented using the HTSeq library (Anders et al. 2015). These tools allow for the analysis of RNA-seq data in terms of sub-exon paths, as in casper (Rossell et al. 2014).
Fusion detection
We used two different fusion callers for the analyses presented in this manuscript. For the patient samples, we used our MI-ONCOSEQ pipeline, which is based on a modified version of TopHat-Fusion (Kim and Salzberg 2011) version 2.0.4, GRCh37 (excluding unplaced contigs), Ensembl 66, with the following nondefault settings “--keep-fasta-order --no-coverage-search --fusion-min-dist 0 --fusion-anchor-length 13 --fusion-ignore-chromosomes chrM.” For the cell line samples, we used FusionCatcher (https://code.google.com/p/fusioncatcher/) 0.99.2b, GRCh37, Ensembl 74, with all default settings.
Variant calling
Picard Tools (http://broadinstitute.github.io/picard) was used to remove duplicates, sort, and index the BAM files. The SplitNCigarReads tool in GATK (Van der Auwera et al. 2002; McKenna et al. 2010; DePristo et al. 2011) version 3.1 was used to split reads spanning splice junctions into exon segments and to hard-clip the sequences overhanging into introns. BaseRecalibrator, HaplotypeCaller, and VariantFiltration from GATK3.1 were used to recalibrate, call variants, and filter the candidates based on Fisher Strand values (FS > 30.0) and Qual By Depth values (QD < 2.0). We further applied filtering steps in SNPiR (Piskol et al. 2013; Ramaswami and Li 2014) to remove mismatches at 5′ read ends, sites in repeat regions (UCSC Genome Browser), and sites in homopolymer runs, and to remove known RNA editing sites. Repeat regions annotated by RepeatMasker were obtained through the UCSC, and the known RNA editing sites were downloaded from the RADAR database. ANNOVAR version-2013-08-23 (Wang et al. 2010) was used for annotation based on gene models from Ensembl.
Data access
The cell line data (40 libraries) generated as part of this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE64113. Patient transcriptomes (30 libraries) are appended to the database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) study numbers phs000554.v1.p1 and phs000567.v1.p1.
Acknowledgments
This work was supported in part by the Prostate Cancer Foundation (PCF351883), the National Institutes of Health S.P.O.R.E. (P50CA69568), a Clinical Sequencing Exploratory Research (CSER) Consortium grant (1UM1HG006508), and an EDRN grant (U01CA111275). M.C. is supported by Young Investigator Awards from the Prostate Cancer Foundation. A.M.C. is supported by the Prostate Cancer Foundation and is an American Cancer Society Professor and A. Alfred Taubman Scholar.
Author contributions: D.R., M.C., and A.M.C. conceived the study and analyses. D.R. and Y.-M.W. developed the capture transcriptome protocol. M.C. performed the data analysis with assistance from M.W., C.B., and R.L. D.R. and Y.-M.W. performed RNA in vitro degradation. D.R., Y.-M.W., F.S., R.W., and X.C. prepared and/or sequenced the RNA-seq libraries. J.S. and R.M. performed prostate autopsies and pathology review. R.C. and D.L. referred the SFT patient and provided case samples and data. D.L. reviewed the SFT pathology. M.C. and A.M.C. wrote the manuscript. All authors discussed results and commented on the manuscript.
Footnotes
-
↵7 Co-senior authors
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.189621.115.
- Received January 14, 2015.
- Accepted July 15, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








