
Effect of minichromosome maintenance protein 2 deficiency on the locations of DNA replication origins
- Dimiter Kunnev1,
- Amy Freeland1,
- Maochun Qin2,
- Robert W. Leach2,
- Jianmin Wang2,
- Rajani M. Shenoy1 and
- Steven C. Pruitt1
- 1Department of Molecular and Cellular Biology;
- 2Department of Biostatistics and Bioinformatics, Roswell Park Cancer Institute, Buffalo, New York 14263, USA
- Corresponding author: steven.pruitt{at}roswellpark.org
Abstract
Minichromosome maintenance (MCM) proteins are loaded onto chromatin during G1-phase and define potential locations of DNA replication initiation. MCM protein deficiency results in genome instability and high rates of cancer in mouse models. Here we develop a method of nascent strand capture and release and show that MCM2 deficiency reduces DNA replication initiation in gene-rich regions of the genome. DNA structural properties are shown to correlate with sequence motifs associated with replication origins and with locations that are preferentially affected by MCM2 deficiency. Reduced nascent strand density correlates with sites of recurrent focal CNVs in tumors arising in MCM2-deficient mice, consistent with a direct relationship between sites of reduced DNA replication initiation and genetic damage. Between 10% and 90% of human tumors, depending on type, carry heterozygous loss or mutation of one or more MCM2-7 genes, which is expected to compromise DNA replication origin licensing and result in elevated rates of genome damage at a subset of gene-rich locations.
During the G1-phase of the cell cycle, locations at which replication will be allowed to initiate in the subsequent S-phase are established by the process of replication origin licensing in which the origin recognition complex (ORC), CDC6, and CDT1 load an inactive form of the minichromosome maintenance (MCM) 2-7 hexameric helicase onto chromatin to form pre-replication complexes (Masai et al. 2010). Unlike other components of the pre-replication complex that bind only transiently, once MCM proteins are loaded onto the chromatin, their association is irreversible until they are released during DNA replication in S-phase (Kuipers et al. 2011). In late G1, increasing activities of CDKs and DBF4-dependent CDC7 kinase, in conjunction with inhibition of CDT1 by geminin, prevent additional loading of MCM proteins, and this restriction remains in place until mitosis (Diffley 2004). Hence, the locations at which MCM protein complexes are bound during G1-phase define locations at which replication can subsequently initiate through the activation of their helicase activity during S-phase.
In many cases, cells license an excess of potential DNA replication origins by loading MCM protein complexes above the number required for chromosomal replication (Dimitrova et al. 1999; Hyrien et al. 2003). A variety of studies have shown that these excess, normally dormant origins can become active under conditions in which nearby replication forks have stalled. The licensing of dormant origins is postulated to serve as a backup system to allow completion of replication in the event of replication fork stalling (Woodward et al. 2006; Ge et al. 2007; Ibarra et al. 2008). The importance of sufficient DNA replication origin licensing to the maintenance of genome stability is demonstrated by the high rates of cancer incidence in mice in which MCM levels or function are reduced (Pruitt et al. 2007; Shima et al. 2007; Kunnev et al. 2010; Kawabata et al. 2011; Rusiniak et al. 2012). Further, under conditions in which the demand for cell proliferation is high, or where cells are transitioning from a low to an accelerated rate of growth, MCM levels have been shown to be limiting, resulting in a reduced level of primary or dormant origin licensing (Orr et al. 2010).
Here we use nascent strand analysis to determine if reduced MCM levels result in measureable changes in DNA replication origin usage. To identify sites of DNA replication initiation, previous studies have used newly replicated nascent DNA strands that are size selected for those sufficiently short to lie near to origins of replication but not so short that they include Okazaki fragments (short nascent strands [SNS]). A widely used method for preparation of SNS is size fraction of denatured genomic DNA using sucrose gradients (Vassilev and Johnson 1989). However, such preparations are heavily contaminated with broken fragments of genomic DNA, resulting in an unacceptably high background. One approach to removing these contaminating fragments takes advantage of lambda exonuclease's ability to catalyze 5′ to 3′ degradation of DNA but not RNA (Bielinsky and Gerbi 1998). Hence, in principle, nascent DNA strands are protected from degradation by their 5′ RNA leader, whereas any contaminating broken strands of DNA are not. This method has been used in large-scale analysis of replication origins in human (e.g., Cadoret et al. 2008; Karnani et al. 2010; Besnard et al. 2012) and mouse (Cayrou et al. 2011) cells using tiling arrays or next-generation sequencing (NGS) to localize SNS within defined regions of the genome. A recurrent observation from studies based on SNS prepared using lambda exonuclease is that sequences from CpG islands, and GC-rich regions of the chromosome generally, are enriched.
Despite the overall agreement between the various data sets, there are concerns about the accuracy and interpretation of the SNS distributions derived from lambda exonuclease-based analyses. Lambda exonuclease is inefficient in digesting single-strand DNAs and some studies (e.g., Cayrou et al. 2011) have used repeated rounds of digestion to attempt to minimize background. Further, it is known that GC-rich sequences are particularly resistant to digestion by lambda exonuclease (Perkins et al. 2003), raising the concern that this property of the nuclease could result in overrepresentation of these sequences.
Here we develop an alternative method for purification of SNSs-based size fractionation of denatured genomic DNA, 5′-biotinylation, capture on a streptavidin substrate, and specific release of nascent strands by ribonuclease digestion of the 5′ RNA leader. We refer to this method as nascent strand capture and release (NSCR). We used next-generation sequencing (NGS) of SNSs prepared by NSCR to compare origin usage in wild-type (wt) and MCM2-deficient mouse embryonic fibroblasts (MEFs).
Results
Identification of DNA replication origins by nascent strand capture and release (NSCR)
The NSCR method is based on a positive selection for the ∼12-nt RNA leader that is introduced at the 5′ end of nascent strand DNA by primase activity during the initiation of DNA replication. The ability of primase to create chimeric RNA:DNA molecules is a property of this enzyme, and DNA molecules containing an RNA leader are a signature of DNA replication. We utilize this property as a positive selection for nascent DNA strands, as shown in Figure 1A. As for prior methods, genomic DNA from proliferating cells is heat denatured and fractionated based on size sedimentation on a sucrose gradient. Material recovered from the gradient contains both nascent DNA strands and DNA fragments resulting from breakage of genomic DNA. These molecules are then 5′ biotinylated using maleimide chemistry and bound to a streptavidin column. After extensive washing, nascent DNA strands are, specifically, released from the column by digesting the 5′ RNA leader using RNase I. RNase I is chosen for this step due to its lack of sequence specificity (Meador et al. 1990).
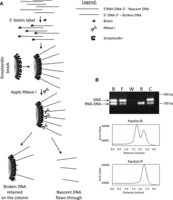
Isolation of nascent strands by nascent strand capture and release (NSCR). (A) Schematic of the approach in which a mixture of RNA-DNA chimeric nascent strands and similar sized contaminating DNA strands are first 5′ end labeled by addition of a 5′ thiophosphate and chemical modification with biotinylated maleimide. 5′-biotinylated oligonucleotides are then bound to a streptavidin column, and the nascent strand component of the bound molecules are specifically released using RNase I. (B) The efficacy of the method is demonstrated by using known substrates in which a mixture of a DNA fragment (representing contaminating DNA) and an RNA-DNA chimera (representing nascent strands and generated as an amplicon from genomic DNA using synthetic RNA[12 nt]-DNA[20 nt] primers) were mixed, 5′ biotinylated, and separated using the methodology. Lanes: B shows the input ratio; F shows the flow through following binding to the column; W shows mock treated fraction after the final wash; R shows material released by RNase I; and C shows material remaining on the column following RNase I treatment as recovered by alkali treatment. Densotometric tracings for lanes B and R are shown below the gel image.
The efficacy of the NSCR approach was first tested on an artificially created mixture of chimeric RNA:DNA and DNA molecules representing nascent strands and contaminating genomic fragments, respectively. The mixture was biotinylated, loaded to a streptavidin column, and processed for nascent strand isolation, in which samples of the biotinylated starting material (B), flow through (F), final wash (W), RNase I released (R), and material remaining bound to the column following RNase I release (C) were assayed using multiplexed PCR with primers specific to either the chimeric RNA-DNA or DNA molecules (Fig. 1B). This control study demonstrates that although there is some loss of chimeric molecules due to failure to bind initially and failure to be released from the column by RNase I, the material that is released is enriched by a factor of >50 based on densitometry of the PCR amplicons.
NSCR analysis of mouse embryonic fibroblast (MEF) DNAs
NSCR was used to compare short nascent strands from wt or MCM2-deficient MEFs. Despite the reduction of dormant origins in MCM2-deficient MEFs (Kunnev et al. 2010), the rate of division of these cells is unaffected (Supplemental Fig. S1A–C). NSCR was performed as described in Methods, and the yield of RNase I released material from ∼1 × 108 cells was ∼1–2 ng of single-stranded DNA or ∼20% of the expected yield of ∼5–10 ng (Hamlin et al. 2010). Sufficient DNA for next-generation sequencing was generated by whole-genome amplification (WGA) of the RNase I released fraction resulting from NSCR, except that an aliquot of unamplified material was saved as a control for the effect of the WGA step on the distribution of sequences. Libraries were prepared using NEBNext ChIP Seq Library Prep kits and sequenced using paired-end sequencing on an Illumina HiSeq 2000 platform in the high output mode. In this first experiment (exp. 1) a total of 142.6 × 106 and 105.0 × 106 uniquely mapped sequences were obtained for samples from wt and MCM2-deficient MEFs, respectively. In a repeat of this experiment (exp. 2), NSCR was used to assay SNSs from ∼1 × 108 wt or MCM2-deficient MEFs as previously, except that libraries were prepared for NGS using IntegenX PrepX DNA ChIP Library Prep kits and sequenced on a HiSeq 2000 platform in the rapid sequencing mode. A total of 323 × 106 and 281 × 106 uniquely mapped sequences were obtained for samples from wt and MCM2-deficient MEFs, respectively. For comparison, we generated a total thymic DNA library using an IntegenX Genomic Seq Library Prep kit and sequenced it on an Illumina HiSeq 2000 platform using paired-end sequencing in the high output mode.
Wiggle files representing the frequency with which sequences from NSCR preparations are found at various locations across the genome were prepared and viewed using the UCSC Genome Browser (Kent et al. 2002). Figure 2A shows representative data centering on the Hoxb4 gene over an ∼45-kbp region of Chr 11 and in comparison to results from a previous study (Cayrou et al. 2011), in which nascent strands were prepared using the lambda exonuclease (lambda-exo) method. There is general agreement in the overall distribution of signal from lambda-exo and each of the NSCR-prepared SNS samples over much of this region. However, the NSCR-prepared samples result in more sharply resolved peaks. For example, the region indicated by the bar under track 3 appears as a series of discrete peaks in NSCR data (Fig. 2A, tracks 3–6) but as a single broadly distributed peak in lambda-exo data (Fig. 2A, track 2). There is good concordance between all four of the NSCR SNS data sets (Fig. 2A, tracks 3–6). Genome-wide, for the largest 20% of peaks present in wt exp. 1, 89.6%, 91.7%, and 86.1% overlap with peaks present in the largest 20% of those found in the MCM2-deficient exp. 1, wt exp. 2, and MCM2-deficient exp. 2, respectively (P < 1 × 10−16 in all cases by the hybrid one at limit case test) (Huen and Russell 2010). However, the distribution of sequences between NSCR data from biologically different samples processed in parallel appears more similar than that of biologically equivalent samples processed in different experiments. This observation suggests that differences in sample processing between the experimental groups significantly affected the representation of particular sequences. Comparison of each of the samples across Chr 11 (Fig. 2B) is consistent with this interpretation. Signal from the NSCR wt sample from experiment 1 (Fig. 2B, track 3) is enriched in GC-rich (Fig. 2B, track 1) regions relative to the NSCR wt sample from experiment 2 (Fig. 2B, track 5). Prior studies have shown that amplification bias during library preparation can result in a GC-rich bias (Aird et al. 2011), and since the library preparation methods differed, such a bias could be responsible for the differences between biologically equivalent samples in experiments 1 and 2. Chromosome-wide correlations between data sets are also consistent with the idea that differences in sample preparation skewed the result between experiments 1 and 2. For example, for Chromosome 11, the correlation between wiggle files derived from wt versus MCM2-deficient samples is 0.94 in experiment 1 and 0.80 in experiment 2. However, comparing wiggle files between biologically equivalent samples between the two experiments, which should correlate even more closely, results in correlations for Chr 11 of 0.50 and 0.65 (and 0.755 and 0.55 genome-wide) for wt exp. 1 versus wt exp. 2 and MCM2-deficient exp. 1 versus MCM2-deficient exp. 2 samples, respectively, consistent with a large contribution of sample preparation or sequencing methods to differences between the two experiments.
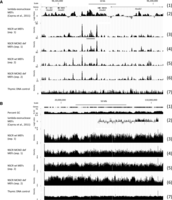
Comparison of nascent strand densities on Chr 11 from lambda exonuclease and NSCR purifications. A comparison of the nascent strand densities obtained by lambda exonuclease-based purification from wt MEFs in a previous study (Cayrou et al. 2011) and by NSCR from wt or MCM2-deficient MEFs in the present study. (A) An ∼45-kbp region of Chr 11 centered on the Hoxb4 gene; (B) all of Chr 11. For each panel: (1) scale and gene tracks; (2) lambda exonuclease SNS data; (3,5) NSCR-SNS data from wt cells, experiments 1 and 2; (4,6) NSCR-SNS data from MCM2-deficient cells, experiments 1 and 2; (7) total genomic DNA from thymus. In B, percentage GC is included in (1).
To examine the distribution of SNS sequences prepared by NSCR prior to genome amplification, we used PCR assays. Regions of the genome showing various peak heights were identified from NGS data, and PCR primers were designed within these regions. In three of four cases examined (Supplemental Figs. S1F,G), similar peaks were present in NSCR-SNS data from both experiment 1 and 2, and the signal level from PCR corresponds to the peak height in each of these cases. In one case (Supplemental Fig. S1H), the NSCR-SNS peak assayed by PCR was more abundant in the data set from experiment 1 relative to experiment 2 but, nonetheless, showed products in the PCR assay.
To confirm that DNAs enriched by NSCR are derived from nascent strands, we used a line of mice in which cell proliferation can be suppressed by doxycycline-dependent overexpression of CDKN1B (p27kip1) (Pruitt et al. 2013). BrdU labeling of MEFs derived from this line is reduced from 11.5% in untreated cells to 0.8% when treated with doxycycline (Supplemental Fig.S1D,E). SNSs recovered using NSCR from untreated and doxycycline treated cells are also reduced, as assayed by PCR at peak positions described above (Supplemental Fig. S1F–H), paralleling the reduction in proliferation (Supplemental Fig. S1I–K).
Effect of MCM2-deficiency on nascent strand distribution
To gain a better understanding of the nature of the regions at which nascent strands are preferentially reduced in MCM2-deficient cells, we normalized wiggle files representing nascent strand sequences obtained from NSCR of wt and MCM2-deficient MEFs. We then subtracted the number of sequences present at each location across the genome of MCM2-deficient cells from the value at each corresponding location of wt cells. Difference files for both experiments 1 and 2 were generated and are displayed on the UCSC Genome Browser for Chr 11 (Fig. 3A, tracks 1 and 2). This approach normalizes many of the differences resulting from changes in the library preparation and NGS steps between experiments 1 and 2 and better represents the consequences of MCM2 deficiency on SNS abundance. These results show that despite the differences between experiments 1 and 2, the effects of MCM2 deficiency are reproduced. For example, the correlation between the difference files for Chr 11 (Fig. 3A, tracks 1 and 2) is 0.88, and genome-wide, this value is 0.917.
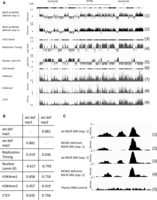
Effect of MCM2 deficiency on nascent strand density. (A) All of Chr 11 (assembly mm9) with scale and chromosome band above the tracks. (1) Wt-deficient difference for exp. 1; (2) wt-deficient difference for exp. 2; (3) UCSC gene density; (4) replication timing (FSU repli-ChIP for MEF; The ENCODE Project Consortium 2012, FSU ENCODE group); (5) lamin B1 (NKI nuclear lamina, lamin B1 for MEF; Peric-Hupkes et al. 2010); (6) CpG island density; (7) H3K4me1 (LICR histone H3K4me1 for MEF; The ENCODE Project Consortium 2012, Ren Laboratory); (8) H3K4me3 (LICR histone H3K4me3 for MEF; The ENCODE Project Consortium 2012, Ren Laboratory); (9) CTCF (LICR TFBS CTCF for MEF; The ENCODE Project Consortium 2012, Ren Laboratory). (B) Correlation coefficients between different tracks calculated using the UCSC Table Browser for Chr 11 as indicated in the figure. C shows a ∼2-kbp region from Chr 10 where (1) is wt exp. 1; (2) is MCM2-deficient exp. 1; (3) is wt exp. 2; (4) is MCM2-deficient exp. 2; and (5) is total thymic DNA for the same region (control).
Comparisons of the difference file tracks with tracks for gene density (Fig. 3A, track 3), replication timing (Fig. 3A, track 4), nuclear lamin B1 (Fig. 3A, track 5), CpG islands (Fig. 3A, track 6), H3K4me1 (Fig. 3A, track 7), H3K4me3 (Fig. 3A, track 8), and CTCF (Fig. 3A, track 9) suggest that MCM2 deficiency results in a preferential reduction of nascent strands within gene dense, early replicating, regions of the genome. The strongest associations are with proteins mediating nuclear architecture, including a negative correlation with lamin B1 and a positive correlation with CTCF (Fig. 3B). In addition to domain-wide differences, MCM2 deficiency also differentially affects origin strength locally. For example, Figure 3B shows an ∼2-kbp region of Chromosome 10 containing three SNS peaks in wt cells where the center peak is reduced in MCM2-deficient cells while the peaks to the left and right are much less affected. Results such as these suggest that local factors can differentially affect MCM protein complex loading during origin licensing.
Sequence elements affecting MCM2 concentration-dependent origin site selection
Sequence elements that have been implicated in defining DNA replication origins in prior studies were examined to determine if their presence correlates with a differential effect of MCM2 deficiency on SNS peak height. Prior studies of SNSs recovered from human cells using the lambda exonuclease method have shown that 67% of SNS peaks are associated with DNA sequences with G-quadruplex potential (G3N1-7)4, where a peak was considered associated if a G-quadruplex sequence is located within 2 kbp to either side (Besnard et al. 2012). The SNS peaks identified here using NSCR are also associated with G-quadruplex, (G3N1-7)4, forming sequences where 49.4% and 48.4% of the largest 5% of the peaks from wt and MCM2-deficient cells, respectively, are found with 2 kbp of such a sequence. However, plotting the distribution of distances from the peak maxima to the G-containing strand of the G-quadruplex sequences (Fig. 4A,B) shows that a subset of <6% of SNS peaks exhibit a tight and orientation-specific relationship with G-quadruplex sequence elements (Supplemental Fig. S2) with a maxima located 186 bp 3′ to the center of the G-quadruplex. Despite their location within 2 kbp to either side of SNS peaks, the remaining quadruplex sequences appear randomly distributed. Further, only ∼5.2% of G-quadruplex sequences are tightly associated with SNS peaks (data not shown). These observations suggest that although the presence of sequences with G-quadruplex-forming potential could play a strong role in defining the precise location of a subset of origins, this sequence is neither necessary nor sufficient to define most origins, at least in the mouse cells studied here. Further, the proportion of SNS peaks that are tightly associated with a G-quadruplex element is similar between samples from wt and MCM2-deficient cells.
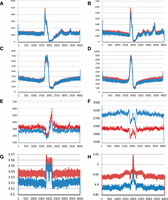
Sequence motifs enriched near to NSCR-SNS peaks. (A–D) Plots of the number of instances (y-axis) that the midpoint of G-quadruplexes (A,B) or TGn ≥ 4 dinucleotide repeats (C,D), oriented relative to the G or TG-containing strands, respectively, occur relative to the location of SNS peak maxima (position 2001) for 4-kbp regions of DNA surrounding each peak. Data from wt cells are shown in red and MCM2-deficient cells in blue, in which A and C show results from exp. 1, and B and D show results from exp. 2. (E,F) Similar plots of motifs enriched in wt unique (E; CYCAGCC) or MCM2-deficient unique (F; ATAWTW) peaks from exp. 1 (wt: red; MCM2-deficient: blue). Plots for additional motifs and for exp. 2 peaks are shown in Supplemental Figures S3 and S4. (G,H) Structural properties of DNA plotted relative to NSCR-SNS peak positions, in which G is average DNA stiffness and H is average consensus DNA bendability for wt (red) and MCM2-deficient (blue) “unique” peaks for peaks that are common between exp. 1 and exp. 2.
TG dinucleotide repeats have been associated with DNA replication origins in Drosophila cells (Cayrou et al. 2011). Similar to the case for G-quadruplex elements, plots of the distribution of distances from SNS peak maxima to the midpoint of the TG-containing strand of (TG)n ≥ 4 elements (Fig. 4C,D) demonstrate that a subset of SNS peaks are tightly associated with TG dinucleotide repeats, where the maxima is 165 bp 5′ to the SNS peak. Further, <9% of SNS peaks are within the 260 bp 3′ to a (TG)n ≥ 4 element (Supplemental Fig. S2). Similar to the case for G-quadruplex elements, there is little difference between wt and MCM2-deficient cells in the proportion of peaks that are tightly associated with (TG)n ≥ 4 elements.
To identify sequences that are associated with peaks that are preferentially sensitive to MCM2 deficiency, we first identified peaks that are “unique” to the largest 5% of total SNS peaks from the wt cell data set (i.e., absent from the largest 5% of peaks from the MCM2-deficient cell data set). We then identified overrepresented sequence motifs relative to those present in peaks that are unique to the largest 5% of total peaks from the MCM2-deficient cell data set using DREME (Bailey 2011). Conversely, we also identified sequence motifs overrepresented in the largest 5% of MCM2-deficient cell “unique” SNS peaks. Although the peaks are unique within the 5% cutoff data sets, in most cases they are not absent but rather reduced to a value at which they no longer fall within the top 5% of peaks.
Sequence motifs that were most highly enriched in the peaks “unique” to wt cells are shown in Supplemental Figure S3A (exp. 1), Supplemental Figure S5 (exp. 2), and Supplemental Figure S6 (peaks common to both experiments). The motifs range between 5 and 9 nt (where 6 nt or longer are shown) and exhibit no apparent consensus. The distribution of distances from the peak maxima relative to the midpoint of several of the motifs is plotted in Figure 4E and Supplemental Figures S3B–J and S7A–C. Similar to G-quadruplex and TG dinucleotide repeats, most of these motifs are enriched ∼180 bp to one side of the peak maxima with a corresponding underrepresentation to the other side. This observation suggests that these elements may serve a similar role to that of G-quadruplex or TG dinucleotide repeats, but may allow less efficient MCM protein complex recruitment, resulting in preferential loss of ori function near to these sites in MCM2-deficient cells.
Sequence motifs that are most highly enriched in the peaks “unique” to MCM2-deficient cells are shown in Supplemental Figure S4A (exp. 1), Supplemental Figure S5 (exp. 2), and Supplemental Figure S6 (peaks common to both experiments). These sequences include motifs that overlap with the G-quadruplex and (TG)n ≥ 4 motifs identified previously and which are located ∼165–185 nt from the position of the SNS peaks. Additional motifs (e.g., ATDCATA, ATATGD, and CATAWW) that do not share sequence similarity with G-quadruplex or TG dinucleotide repeats but are preferentially located ∼180 bp from the SNS peak positions are also enriched in peaks “unique” to MCM2-deficient cells. Other motifs (e.g., AAAWWAT and ATAWTW) are not enriched at any particular site, but rather are excluded from positions ∼180 and/or ∼50 bp from SNS peak maxima despite their general overrepresentation in the vicinity of peaks “unique” to MCM2-deficient cells (Fig. 4F; Supplemental Fig. S4E–J).
Based on the idea that a shared structural property accounts for the observation that a variety of different specific sequence motifs localize to a position ∼180 bp from SNS peak positions, we examined DNA stiffness (Gromiha 2000) and consensus bendability (a measure of AT and GC type intrinsic curvature propensity) (Vlahovicek et al. 2003) of the motifs. Motifs enriched in either wt “unique” or MCM2-deficient “unique” peaks that are overrepresented at positions 160–185 exhibit values for consensus DNA bendability that are well above average (Supplemental Figs. S3, S4, S6, blue highlight) (average consensus DNA bendability is ∼4.95 for DNA within 2 kbp of total peaks from wt cells). Conversely, motifs that are preferentially excluded in this region exhibit consensus bendability that is well below average (Supplemental Figs. S4, S6, yellow highlight). In addition, motifs with low consensus DNA bendability scores tend to be excluded from a position ∼50 bp from SNS peak maxima. These observations suggest that high intrinsic DNA curvature at a position between 165 and 185 bp, and to a lesser extent at a position 50 bp, from the site of replication initiation is favorable for replication origin licensing.
To examine the contribution of DNA structure to determining the effect of MCM2 deficiency on origin usage, we calculated the average consensus DNA stiffness (Fig. 4G) and DNA bendability (Fig. 4H) for each position between −2000 and +2000 bp, relative to SNS peaks, for common peaks between exp. 1 and exp. 2 but that are “unique” to wt or MCM2-deficient samples (95% cutoff). Results from these studies show a correlation between DNA structural properties (within ∼180 bp) and the position of nascent strand peaks in both wt and MCM2-deficient cells. However, peaks that are sensitive to MCM2 deficiency (i.e., wt “unique” peaks) tend to occur in regions that have overall higher values in both DNA stiffness and consensus bendability, particularly within ∼500 bp of the SNS peak maxima.
The distribution of sequence motifs that are differentially enriched near to MCM2-sensitive or MCM2-insensitive peaks across large domains in the genome correlates well with domain-wide differences in nascent strand density between wt and MCM2-deficient cells. For example, Supplemental Figure S7G shows an ∼4-Mbp region of Chr 10 containing two flanking regions in which SNSs are preferentially lost in MCM2-deficient cells and a central domain of ∼500 kbp in which SNS are less affected by MCM2 deficiency (wt minus MCM2 deficiency, track 1). Tracks 2 and 3 show the distributions of two motifs that are enriched in wt “unique” peaks, and tracks 4 and 5 show the distributions of two motifs that are enriched in MCM2 deficiency “unique” peaks. Tracks showing CpG islands (6) gene density (7) and replication timing (8) are also included. These results suggest that much of the differential effect of MCM2 deficiency on origin usage in gene dense, early replicating regions can be accounted for by their DNA sequence composition.
Locations of reduced nascent strand density correlate with recurrent copy number variations (CNVs) in tumors resulting from MCM2 deficiency
MCM2-deficient mice on a 129Sv genetic background develop thymic lymphocytic leukemia (T-LL) with early onset (average age 12 wk) and complete penetrance (Pruitt et al. 2007; Kunnev et al. 2010). The genetic lesions occurring in these tumors have been characterized and are predominately focal deletions averaging ∼450 kbp in length (Rusiniak et al. 2012). To examine the possibility that origin usage in MEFs is reduced near locations where deletions are found in tumors of MCM2-deficient mice, we compared the NSCR wt minus MCM2-deficient wiggle file in the UCSC Genome Browser to locations of the previously identified deletions from tumors arising in MCM2-deficient mice (e.g., Supplemental Fig. 5A,B). This comparison shows that many of the most frequent deletion sites correlate with the locations where MCM2 deficiency results in the largest reduction in nascent strands. Differences in nascent strand density were determined for all deletion events identified in tumors from MCM2-deficient mice and are plotted in rank order (Fig. 5C). Of 142 events, 106 showed a reduction in nascent strand density in MCM2-deficient, relative to wt, MEFs over the interval that is deleted in T-LL tumors. Further, with a major exception of the Pten locus on Chr 19, the reduction is greater at locations of recurrent deletions (16/19 recurrent deletion sites show preferential reduction of nascent strands in MCM2-deficient MEFs) (Fig. 5D). ChIP analysis (Supplemental Fig. S8) additionally shows that at locations exhibiting a preferential reduction in nascent strands in MCM2-deficient MEFs (near to Mbd3/Tcf3), there is a corresponding loss of MCM2 binding relative to a location that is less affected (near to Pten).
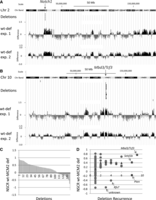
Relationship between the effect of MCM2 deficiency on nascent strand density in MEFs and locations of CNVs in tumors. (A) Region of Chr 2 where recurrent deletions were found in the Notch1 gene (Rusiniak et al. 2012) of thymic lymphocytic leukemias (T-LLs) arising in MCM2-deficient mice as indicated and compared to NSCR wt minus deficient difference values over the same region. (B) Similar comparison for the Mbd3/Tcf3 locus on Chr 10. (C) Plot, in rank order, of the average NSCR wt minus MCM2-deficient values from exp. 1 over the length of each deletion for each of 142 deletions identified in T-LLs arising in MCM2-deficient mice (Rusiniak et al. 2012). (D) The data in D are based on the same deletions as in C, where NSCR wt minus MCM2-deficient values for deletions recurring at the same sites in different tumors were averaged, and the average NSCR wt minus MCM2-deficient value is plotted against the frequency with which deletions recurred (circles). The triangles indicate the average NSCR wt minus MCM2-deficient value for all deletions at different levels of recurrence except that three outliers (Pten, Rfx7, and unknown) were excluded. Similar results were found for data from exp. 2; and for both exp. 1 and exp. 2, bootstrap analysis (Supplemental Fig. S9), and demonstrates a significant (P < 0.001) association between sites of recurrent deletions as well as a preferential effect of MCM2 deficiency on nascent strand density over the deletion intervals.
Discussion
Nascent strand mapping
Nascent strand mapping in mammalian cells has been utilized to localize origins over large regions of the genome in several prior studies (Cadoret et al. 2008; Karnani et al. 2010; Cayrou et al. 2011; Besnard et al. 2012). In these studies, a key step is removal of contaminating broken DNA strands by lambda exonuclease. In principle, this step provides enrichment of nascent DNAs since they are protected from digestion by a 5′ RNA leader. However, the efficiency of lambda exonuclease in digesting single-stranded DNAs is low (Sriprakash et al. 1975), and GC-rich sequences suppress its activity (Perkins et al. 2003). Despite protocols designed to optimize this step (Cayrou et al. 2011), there remains a concern that these preparations are contaminated by DNAs that are not derived from nascent stands. This concern is elevated by the GC-rich nature of sequences recovered by lambda exonuclease-based nascent strand purifications and the observation that such sequences are not strongly correlated with the locations of origins defined by alternative methods (Mesner et al. 2013). In the present study, we have developed a method for enrichment of nascent strands based on a positive selection for the presence of a 5′ RNA leader. Capture of nascent DNA strands by 5′ biotinylation and release with RNase I is not expected to result in enrichment of GC-rich sequences.
Properties of DNA replication origins
Prior studies using lambda exonuclease enriched SNSs have shown that an origin G-rich consensus element (OGRE), CpG islands, and G-quadruplex forming sequences (that are frequently associated with OGRE and CpG islands) are enriched in 2 kbp of DNA surrounding SNS peaks where these elements are present in ∼70% of these regions (Cadoret et al. 2008; Karnani et al. 2010; Cayrou et al. 2011; Besnard et al. 2012). Further, when nascent strand signal strength (Cayrou et al. 2011), a measure of nascent strand density across the regions surrounding specific sequence elements, was plotted, a peak was found at position 280 bp 3′ to the OGRE or G-quadruplex elements. Similar analyses here confirm that G-quadruplex elements are associated with NSCR-SNS peaks with a sharp maxima 186 bp 5′ to a subset of NSCR-SNS peaks. However, at least in the mouse cells utilized in the present study, <6% of NSCR-SNS peaks exhibit this tight association. Further, we have shown that a variety of additional DNA sequence motifs exhibit enrichment at a discrete distance from NSCR-SNS peaks. For example, TG dinucleotide repeats show orientation-specific association with ∼7%–8% of NSCR-SNS peaks with a maximum at ∼165 bp 5′ to the peaks. TG dinucleotide repeats have been associated with replication origins defined by lambda exonuclease SNS analyses of Drosophila cells in a previous study (Cayrou et al. 2011). Similarly, multiple additional sequence motifs are identified in the present study that localize between 160 and 190 bp from NSCR-SNS peaks.
Although the various sequence motifs exhibiting enrichment between 160 and 190 bp from NSCR-SNS peaks do not share a consensus sequence, they do share common structural properties. They exhibit high values for DNA stiffness and consensus DNA bendability, where the latter is considered a measure of intrinsic DNA curvature (Gabrielian et al. 1996; Vlahovicek et al. 2003). Further, sequence motifs showing low consensus bendability values tend to be excluded from this location in replication origins generally. These results are similar to the prior observation that a region of DNA stiffness is located 160 bp 5′ to yeast ARS elements (Chen et al. 2012).
We speculate that DNA structural properties within ∼160–190 bp from the NSCR-SNS peak positions affect binding of components of the DNA replication licensing machinery. Prior studies have shown that in budding yeast, the origin recognition complex (ORC) protects 48 bp of DNA from DNase I digestion, and that this footprint extends to nearly 80 bp when CDC6 binds (Speck and Stillman 2007). Further, DNA contained within the ORC-CDC6 complex exhibits a strong bend such that CDC6 interacts with DNA on either side of the complex. Structural studies suggest that the ORC-CDC6 complex undergoes a transition on loading CDT1-MDM2-7 such that the length of DNA covered by the ORC-CDC6 portion of the complex is reduced to only ∼30 bp, and the CDT1-MCM2-7 portion interacts with only ∼45 additional bp of DNA (Sun et al. 2013). However, if the ORC-CDC6 complex is positioned immediately adjacent to the MCM protein complex, the maximum region of DNA that would be contacted by known components of the replication licensing complex would extend at most 125 bp from the SNS peak, inconsistent with the location of an interaction site 160–190 bp away. One explanation for this discrepancy is that an additional, as yet unidentified, protein interacts at this location (Cayrou et al. 2011). Alternatively, it has been shown for S. pombe (Gaczynska et al. 2004) and Drosophila (Remus et al. 2004) that following the initial binding, DNA is wrapped around the ORC complex, which could result in a distance of ∼160–190 bp between an initial ORC binding site and the location at which the CDT1-MCM2-7 complex is bound. Alternatively, MCM2-7 complex loading has been studied only on naked DNA substrates, and it is unclear whether these studies accurately reflect loading to chromatin. A distance of 160–190 bp would be consistent with the binding of ORC-CDC6 to the linker region between nucleosomes on a chromatin substrate followed by recruitment of the CDT1-MCM2-7 complex to an adjacent linker region separated by a nucleosome core.
Effect of MCM2 deficiency on replication origin usage
A key finding of the present work is that not all DNA replication origins are affected equally by MCM2 deficiency. Origins that are most affected tend to be located in gene-dense early replicating regions of the genome, where their locations correlate positively with increased levels of CTCF, H3K4me1, and H3K4me3 and negatively with lamin B1. However, the degree to which an origin will be affected by MCM2 deficiency is also dependent on local sequence composition. All peaks, regardless of their degree of sensitivity to MCM2 deficiency, exhibit regions that have locally high DNA stiffness values at 160–190 bp from the peak maxima. However, other locations in the vicinity of peaks that are resistant to MCM2 deficiency show reduced values, suggesting that increased DNA flexibility at locations other than 160–190 bp from the SNS peak maxima are advantageous for loading the MCM protein complex. One explanation for this result is that increased flexibility of DNA adjacent to the location of ORC-CDC6 binding facilitates the conformation transitions that accompany either the initial CDT1-MCM2-7 recruitment or the binding of the second MCM2-7 hexamer. The distribution of sequence motifs exhibiting high values on the DNA stiffness and consensus DNA bendability scales are generally accurate indicators of where MCM2 deficiency will have the greatest effect on regional SNS density.
MCM2 deficiency and genome instability
Under conditions in which MCM protein levels are reduced (Ge et al. 2007; Ibarra et al. 2008; Kunnev et al. 2010), although not necessarily in cases where MCM protein function is affected by mutation (Kawabata et al. 2011), it has been shown that inter-ori distance is increased under conditions of replication stress even though the rate of fork progression is not affected. These observations support that replication licensing, rather than the rate of fork movement or stalling, is responsible for the elevated genetic damage and cancers seen in MCM2-deficient mice (Pruitt et al. 2007; Kunnev et al. 2010). The present study shows that reduced origin density (and for the subset of locations assayed, MCM2 binding) occurs in discrete domains across the genome where some locations are much more affected than others. Consistent with a role for increased origin licensing in suppressing genetic damage, locations where there is the greatest loss of SNSs are correlated with locations where recurrent focal CNVs are found in T-LLs arising in MCM2-deficient mice (Rusiniak et al. 2012). Loss of the protection afforded by increased origin licensing at the Mbd3/Tcf3 locus, genes which have been implicated in T-LL in mice and humans previously (e.g., Bain et al. 1997; for review, see Rusiniak et al. 2012), and a few additional sites may drive tumorogenesis in these mice. A major exception to this correlation in T-LLs arising in MCM2-deficient mice occurs at the Pten gene. This exception could reflect differences between origin usage in MEFs and T cells. Alternatively, a requirement for loss of PTEN function may be a rate-limiting step in the progression of these cancers.
Replication licensing in human tumors
MCM2-7 genes are thought to serve essential and nonredundant roles. Not surprisingly, homozygous loss of MCM2-7 genes is seldom, if ever, observed in human tumors. In fact, many tumors contain an elevated proportion of replication-competent cells that express MCM2-7, and this property has been useful as a diagnostic marker (for review, see Giaginis et al. 2010). Analysis of TCGA (The Cancer Genome Atlas Network 2012) data demonstrates that alteration of replication licensing factor genes is a frequent occurrence in human cancers. Mutations or gene amplifications in replication licensing factor genes (RLF genes: MCM 2-7, CDC6, CDT1, and ORC1-6) occur in a few percent of leukemias to 20%–40% in some carcinomas (Supplemental Fig. S10A). Between different tumor types, 55%–74% of mutations in MCM2-7 complex members are predicted to have a medium or high functional effect as determined by the mutation assessor score (Reva et al. 2011). Further, 42%–70% of the mutations lie within conserved MCM protein domains (Supplemental Fig. S11).
Despite amplification of a subset of the MCM2-7 genes in some tumors, there is little evidence for coordinate amplification of all of the members of the complex, and it is unclear whether elevated expression of one complex member will lead to increased or reduced function, due to a stoichiometric imbalance. Further, heterozygous losses occur in an even greater proportion of tumors, in which the frequency of such events ranges from ∼10% to >90% depending on cancer type (Fig. 6; Supplemental Figs. S12, S13), and overall, about half of all tumors exhibit heterozygous loss of at least one MCM2-7 gene. These alterations are reflected at the transcript level (e.g., Supplemental Fig. S10B). Alterations in MCM2-7 and CDT1 are more frequent than in other members (ORC1-6, CDC6) of the replication licensing complex. Statistical analyses of genetic changes in human tumors support that haploinsufficiency and triplosensitivity are significant drivers of the cancer phenotype (Davoli et al. 2013). Further, MCM7 has been identified as a haploinsufficient tumor suppressor in human cancers (Davoli et al. 2013). The effects of MCM2 deficiency in the mouse model suggest that the high proportion of human tumors carrying heterozygous loss of one or more MCM2-7 genes may undergo elevated rates of genetic damage that are concentrated to a subset of gene-rich regions of the genome that can, at least in part, be predicted based on DNA sequence composition.
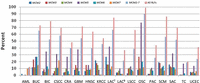
Putative loss-of-function alterations in RLF genes across tumor types. The proportion of tumors that carry heterozygous loss or mutation of MCM2-7 individually or as a group and all RLFs (MCM2-7, CDC6, CDT1, and ORC1-6) as indicated in the key across different cancer types: AML, Acute Myeloid Leukemia; BUC, Bladder Urothelial Carcinoma; BIC, Breast Invasive Carcinoma; CSCC, Cervical Squamous Cell Carcinoma and Endocervical Carcinoma; CRA, Colon and Rectum Adenocarcinoma; GBM, Glioblastoma Multiforme; HNSC, Head and Neck Squamous Cell Carcinoma; KRCC, Kidney Renal Clear Cell Carcinoma; LACT, Lung Adenocarcinoma (TCGA Provisional); LACB (Broad); LSCC, Lung Squamous Cell Carcinoma; OSC, Ovarian Serous Cystadenocarcinoma; PAC, Prostate Adenocarcinoma; SCM, Skin Cutaneous Melanoma; SAC, Stomach Adenocarcinoma; TC, Thyroid Carcinoma; UCEC, Uterine Corpus Endometrioid Carcinoma.
Methods
Cells
For most experiments, passage 4 mouse embryonic fibroblasts (MEFs) from wild-type and MCM2-deficient embryos (Pruitt et al. 2007) were generated as described in Kunnev et al. (2010), and 1 × 108 cells of each genotype were harvested for nascent strand capture and release. For proliferation assays of wt and MCM2-deficient cells, passage 2 MEFs were seeded on a 96-well plate using 6000 cells per well in DMEM supplemented with 10% FBS. At 24 h intervals, wells were taken for an MTT proliferation assay (Sigma-Aldrich Catalog #M5655-1G diluted to 1 mg/mL in completed media and incubated with cells for 1 h at 37°C 5% CO2). After the incubation, media was removed and accumulated MTT was visualized by dissolving in DMSO and measuring absorption at 570 nm. The average and SD for 16 wells was determined. In the experiments shown in Supplemental Figure S1D,E, and I–K, MEFs were derived from R26-M2rtTA;TRE-Tight-Cdkn1b bigenic mice (Pruitt et al. 2013), and cells were either untreated or treated with 10 µg/mL doxycycline for 26 h. DNA was recovered from 1.8 × 108 cells per group for use in NSCR.
Cell cycle analysis by FACS
Cells were treated with 100 µM BrdU for 40 min, trypsinized, washed with PBS and fixed in 70% ethanol. Fixed cells were exposed to 2.0 N HCl and 0.5% Triton X-100 for 30 min, pelleted and resuspended in 100 mM NasB404 pH 8.5, washed with TBP (0.5% Tween 20 and 1% BSA in PBS) and stained first with a 1/200 dilution of anti-BrdU antibody (AXYLL Rat MAB OBT00305) and, following washing with TBP, with a 1/500 dilution of Alexa Fluor 488 goat anti-rat IgG (Life Technologies REF#A11006) and propidium iodide. A minimum of 30,000 events/sample were recorded by FACS.
Nascent strand DNA capture and release (NSCR) of MEF DNA
DNA from ∼1 × 108 wt or MCM2-deficient MEF's was purified using a high salt SDS lysis buffer and proteinase K digestion. At this point, RNA was removed using TRIzol Reagent (Life Technologies) since it would otherwise compete for biotinylation during subsequent steps. DNA was then heat denatured and 300 µg was loaded to each of three sucrose gradients for size fractionation (described in Supplemental Methods). Fractions containing molecules ranging between ∼400 and 2000 nt were identified and pooled. The pooled material was 5′ biotinylated using Vector Laboratories 5′ EndTAq Nucleic Acid Labeling System (MB-9001) with Vector Laboratories biotinylated maleimide (SP-1501) and fractioned using NSCR as described in detail in the Supplemental Methods. Samples of the starting material (B), final wash (W), and RNase I released material (R) were collected. For determination of yield, RNase I released material was converted to double-stranded DNA by random priming and treatment with the Klenow fragment of E. coli DNA polymerase I (Invitrogen 18012-021) and quantified using pico-green staining. This estimate showed that ∼1–2 ng of single-stranded material was recovered per mg of starting genomic DNA. The expected yield of 400–2000 nt nascent strands from 1 × 108 cells is ∼5–10 ng (Hamlin et al. 2010), suggesting a yield of ∼20% of nascent strands by the NSCR method.
Illumina sequencing
For next-generation sequencing, the RNase I released fraction (Fraction R) resulting from NSCR (omitting double-strand conversion with Klenow) was subjected to whole-genome amplification (WGA), except that an aliquot of unamplified material was saved as a control for the effect of the WGA step on the distribution of sequences. Fraction R was amplified using the whole-genome amplification kit WGAII (Sigma-Aldrich; SEQX in experiment 1 and SEQXE in experiment 2) following the provided protocol.
The amplified material was sequenced by the Harvard Medical School Biopolymers Facility using a 50 (exp. 1) or 72 (exp. 2) cycle paired-end protocol on an Illumina HiSeq 2000 platform.
Bioinformatics
A detailed description of the treatment of NGS data from NSCR and other bioinformatic procedures is given in the Supplemental Methods. Data from The Cancer Genome Atlas Project was analyzed using cBioPortal (Cerami et al. 2012).
Data access
NSCR-SNS sequence data from this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA258088. Wiggle files have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession number GSE64933.
Acknowledgments
This work was supported by grants from the NIH-NCI (1R01CA130995), NIH-NIA(R01AG041854), and the Ellison Medical Foundation to S.C.P. Cost of animal maintenance and flow cytometry was supported in part by an NCI-CCS grant to Roswell Park Cancer Institute. The authors acknowledge helpful discussions with Joel Huberman, William Burhans, and Richard Pelroy.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.176099.114.
- Received March 24, 2014.
- Accepted January 26, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








