
Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells
- Monika S. Kowalczyk1,8,
- Itay Tirosh1,8,
- Dirk Heckl2,
- Tata Nageswara Rao3,4,
- Atray Dixit1,
- Brian J. Haas1,
- Rebekka K. Schneider2,
- Amy J. Wagers3,4,5,6,
- Benjamin L. Ebert2 and
- Aviv Regev1,6,7
- 1Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 2Division of Hematology, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 3Harvard Stem Cell Institute and Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, Massachusetts 02138, USA;
- 4Joslin Diabetes Center, Boston, Massachusetts 02215, USA;
- 5Paul F. Glenn Laboratories for the Biological Mechanisms of Aging, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts 02140, USA;
- 7Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02140, USA
- Corresponding author: aregev{at}broadinstitute.org
-
↵8 These authors contributed equally to this work.
Abstract
Both intrinsic cell state changes and variations in the composition of stem cell populations have been implicated as contributors to aging. We used single-cell RNA-seq to dissect variability in hematopoietic stem cell (HSC) and hematopoietic progenitor cell populations from young and old mice from two strains. We found that cell cycle dominates the variability within each population and that there is a lower frequency of cells in the G1 phase among old compared with young long-term HSCs, suggesting that they traverse through G1 faster. Moreover, transcriptional changes in HSCs during aging are inversely related to those upon HSC differentiation, such that old short-term (ST) HSCs resemble young long-term (LT-HSCs), suggesting that they exist in a less differentiated state. Our results indicate both compositional changes and intrinsic, population-wide changes with age and are consistent with a model where a relationship between cell cycle progression and self-renewal versus differentiation of HSCs is affected by aging and may contribute to the functional decline of old HSCs.
A rare population of multipotent hematopoietic stem cells (HSCs) is required for the continuous production of millions of mature blood cells while maintaining a correct balance between the different lineages. At the apex of the hematopoietic hierarchy reside the most primitive long-term reconstituting HSCs (LT-HSCs). LT-HSCs can undergo three types of cell division: (1) a renewal symmetric cell division to produce two LT-HSC daughter cells that replenish the LT-HSC pool; (2) a commitment symmetric division to replenish committed cells producing short-term reconstituting HSCs (ST-HSCs) and, subsequently, multipotent progenitors (MPPs); and (3) asymmetric division, where one daughter cell remains a stem cell while the other becomes committed.
This remarkable capacity of HSCs declines with age as reflected by an accumulation of HSCs in the bone marrow (BM) (Morrison et al. 1996; de Haan et al. 1997; Sudo et al. 2000; Rossi et al. 2005) that shows decreased regenerative potential (Morrison et al. 1996; Sudo et al. 2000; Kim et al. 2003; Rossi et al. 2005; Dykstra et al. 2011) and a myeloid-skewed differentiation potential (Sudo et al. 2000; Kim et al. 2003; Liang et al. 2005; Rossi et al. 2005). Relatedly, in the elderly, there is an increased incidence of myeloid diseases such as leukemias (Lichtman and Rowe 2004), a decreased competence of the adaptive immune system (Linton and Dorshkind 2004), and the onset of anemia (Beghe et al. 2004).
Despite extensive studies documenting the decline of HSC function during aging, the molecular mechanisms underlying HSC aging have remained obscure. Current views of stem cell aging converge to two main models: (1) clonal selection, implying that multiple HSC clones with specific phenotypes coexist but their relative frequencies change with age, and (2) a global population shift in intrinsic cell states, in which all HSCs in the population undergo coordinated changes in functional potential with age. Previous transcriptional profiling limited to populations of young and old LT-HSCs demonstrated up-regulation of myeloid genes and down-regulation of lymphoid and cell cycle genes with age (Rossi et al. 2005; Chambers et al. 2007; Sun et al. 2014). However, such population measurements may obscure important cell-to-cell variability in gene expression and could not definitively distinguish between these two models and thus cannot determine whether there are distinct, cell-intrinsic, functional states of HSCs that are age dependent or whether the observed transcriptional differences between bulk populations reflect changes in the proportions of subgroups of cells.
New advances in single-cell genomics (Wills et al. 2013), especially single-cell RNA-seq (Hashimshony et al. 2012; Ramskold et al. 2012), have opened the way to characterize distinct functional states of individual cells, even within seemingly homogeneous immune cell populations (Shalek et al. 2013, 2014; Mahata et al. 2014). These should allow us to distinguish changes that arise from cell-intrinsic differences in transcriptional states from those that reflect changes in the proportion of subpopulations. Furthermore, single-cell analysis should allow us to relate prospective profiles of HSCs that have just been isolated with known heterogeneity in their retrospective functional capacity in transplantation assays.
Here, we leveraged single-cell RNA-seq to directly assess transcriptional heterogeneity within the HSCs and how it may change with age in the steady-state unperturbed hematopoiesis. Given that HSCs are functionally heterogeneous as revealed through transplantation studies (Dykstra et al. 2007; Challen et al. 2010; Muller-Sieburg et al. 2012), we hypothesized that this retrospective variability (assessed through the outcome of a transplant) would be prospectively reflected in the transcriptional profiles of cells in unperturbed conditions.
Results
Single-cell RNA-seq of approximately 1200 HSCs
To systematically characterize the global transcriptional landscape of individual cells in the course of the first steps of mouse hematopoiesis, we used multiparameter fluorescence-activated cell sorting (FACS) followed by single-cell RNA-seq using SMART-seq, as previously described (Methods) (Ramskold et al. 2012; Shalek et al. 2013, 2014). We prospectively isolated three cell types using LSK (lineage−, SCA1+, KIT+) and SLAM (signaling lymphocyte activation molecule) markers whose expression is conserved across mouse strains and during aging (Kiel et al. 2005; Yilmaz et al. 2006): long-term HSCs (LSK CD150+CD48−), ST-HSCs (LSK CD150−CD48−), and MPPs (LSK CD150−CD48+) from young (2–3 mo) and old (>22 mo) C57BL/6 mice (Fig. 1A–C). Consistent with previous work (Morrison et al. 1996; de Haan et al. 1997; Sudo et al. 2000; Rossi et al. 2005), we observed a significant expansion of LT-HSCs with age (approximately sixfold, P < 0.001) in total BM (Fig. 1D) and in the stem cell compartment (Fig. 1E). In contrast, ST-HSC frequency within the LSK compartment of old mice was comparable and MPP frequency decreased, possibly indicating an imbalance between self-renewal and differentiation.
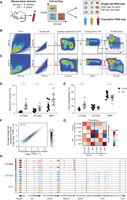
Single-cell RNA-seq of young and old HSCs. (A) Overview of experimental design. (B,C) Sorting strategy for isolating LT-HSCs (LSK CD48−CD150+), ST-HSCs (LSK CD48−CD150−), and MPPs (LSK CD48+CD150−) from young (B) and old (C) C57BL/6 mice. (D,E) LT-HSC compartment expands during aging. Shown are frequencies of LT-HSC, ST-HSC, and MPPs (x-axis) in young (black) and old (white) C57BL/6 mice as a percentage of bone marrow (BM; D) or stem cell compartment (lineage− SCA1+KIT+, LSK; E). Statistically significant differences are as follows: (**) P < 0.01, (*) P < 0.05; n = 8–10. (F) Single-cell RNA-seq recapitulates population RNA-seq. Shown are expression levels for all genes calculated from RNA-seq of a population of young LT-HSCs (x-axis) and by averaging expression levels from approximately 200 single young LT-HSCs (y-axis). The Pearson correlation coefficient (r = 0.9) is denoted. Gray scale bar indicates gene density. (G) Heatmap of Pearson correlation coefficients (r; color bar) between pairs of RNA-seq profiles of populations (columns) and matching averaged single-cell data (rows) from C57BL/6. (H) RNA-seq coverage of known cell surface markers in representative cells from young C57BL/6 mice (plot generated by the Integrative Genome Viewer 2.3) (Robinson et al. 2011; Thorvaldsdottir et al. 2013).
We generated 1152 single-cell RNA-seq profiles, along with corresponding population controls, from six populations: three cell types (LT-HSCs, ST-HSCs, and MPPs) at each of the two ages (2–3 and >22 mo; nearly 200 cells per cell type and age). Our stringent filtering criteria retained 1059 high-quality libraries for subsequent analyses (about 176 per cell type and age on average; for QC details and filtering criteria, see Methods). Aggregated in silico, the average single-cell gene expression profile of the individual cells from a given cell type reproduced the gene expression profile of its matching population control (r ∼ 0.9 on average) (Fig. 1F,G). Additionally, profiles of populations or aggregated single cells were highly correlated between young and old mice, indicating that aging-related differences may be subtle (Fig. 1G). Finally, the RNA expression levels of markers used for sorting these cell types or known to correlate with them reproduced known associations (Fig. 1H).
Cell cycle is a main source of transcriptional variation between HSCs
To uncover the main sources of variation, we performed principal component analyses (PCA) for each cell type and age separately. Strikingly, in every case, the top principal components (PCs) were associated primarily with cell cycle–related genes (Fig. 2A), indicating that transcriptome heterogeneity here is dominated by cell cycle status. Furthermore, analysis of known mouse hematopoietic transcriptional modules (Jojic et al. 2013; Shay et al. 2013) showed strong coregulation across single cells only for genes in cell cycle modules (Supplemental Fig. S1A). When we analyzed only those cells without appreciable expression of cell cycle genes (65% of all cells), the highest remaining coregulation was for a ribosome module, with an average correlation lower than 0.1 in each of the six populations (Supplemental Fig. S1B). We thus concluded that cell cycle reflects the main source of heterogeneity within the transcriptomes of each HSC cell type and age.

Old LT-HSCs have a lower frequency of cells in G1 phase of the cell cycle. (A) The top eight PCs in each cell type and age. Shown is the percentage of annotated cell cycle genes (y-axis) in the top 100 genes that correlate with each of the PCs (x-axis) in each population. (B) Cell cycle analysis on mouse KIT-enriched BM cells stained with pyronin Y (y-axis) and Hoechst (x-axis), reflecting for each cell the amount of RNA and DNA, respectively. Sorting gates and cell cycle phases are indicated. (C) RNA-seq of KIT-enriched BM cells at different cell cycle phases. Shown is the average expression of G1/S genes (x-axis) and G2/M genes (y-axis) from RNA sequenced from gates in B (color-coded). (D) HSC single-cell transcriptomes can be clustered by their cell cycle status. Heatmap shows average expression of cell cycle phases gene signatures (rows) in each cell (column). The cells are partitioned into three clusters expressing the G1/S program, G2/M program, or neither. (E) Cell cycle distribution changes as a function of cell type and age. The percentage of cells in cluster 1 (G2/M, gray) and cluster 2 (G1/S, black), within each cell type, for young (x-axis) and old (y-axis) HSCs. (F) Cell cycle trajectory inferred from single-cell RNA-seq. Shown is the average expression of G1/S genes (x-axis) and G2/M genes (y-axis). The arrow and labels reflect inferred cell cycle progression. (G,H) Lower frequency of G1 cells among old LT-HSCs based on FACS analysis. (G) Shown are cell frequencies in G1 (black) and S-G2-M (gray) in cells from young (x-axis) and old (y-axis) mice based on intracellular staining with Ki67/Hoechst. (H) Representative FACS plots for young and old LT-HSCs from C57BL/6 mice. (I) The frequency of young and old LT-HSCs in S phase based on in vivo bromodeoxyuridine (BrdU) incorporation. (J) Representative BrdU FACS plots for young and old LT-HSCs from C57BL/6 mice.
To further dissect the cell cycle states, we scored each cell for its likely cell cycle phase using signatures for the G1/S, S, G2, and G2/M phases defined based on functional annotations (The Reference Genome Group of the Gene Ontology Consortium 2009) and profiles from synchronized HeLa cells (Whitfield et al. 2002) and further refined by coexpression in our profiles (Methods). To directly confirm that these signatures correspond to different cell cycle phases, we separated cell cycle phases of KIT-enriched cells by concurrent pyronin Y and Hoechst staining (Fig. 2B). RNA sequenced from 11 gates representing the cell cycle continuum showed precise partitioning based on our signatures as expected (Fig. 2C). Lastly, we confirmed that these signatures robustly detect cell cycle phases in other single-cell data sets from human and mouse (Supplemental Fig. S2; Macosko et al. 2015).
Clustering all HSCs by the expression of these signatures uncovered three distinct groups of cells: cluster 1, which expresses primarily G2/M-phase genes (e.g., mitosis genes); cluster 2, with high expression of G1/S-phase genes (e.g., DNA replication genes); and cluster 3, which does not show expression of cell cycle genes (noncycling or quiescent cells) (Fig. 2D; Supplemental Fig. S3B). The frequency of cells in the G1/S and G2/M clusters (cycling cells) was considerably higher in MPPs than in LT- and ST-HSCs (Fig. 2E; Supplemental Fig. S3C–E), consistent with the known high level of proliferation of MPPs (Wilson et al. 2008).
Almost a third of the analyzed genes (2708 genes, ∼32%) were found to have significant cell cycle–dependent expression changes that were consistent among multiple HSC types or ages (Supplemental Fig. S4A; Supplemental Table S1). The vast majority of cell cycle–regulated genes (92%) were up-regulated in cycling versus noncycling cells (for phase-specific up-regulation, see Supplemental Table S1); the minority of cell cycle–regulated genes (8%) that were up-regulated in noncycling versus cycling cells included several transcription factors that could be important for regulating HSC quiescence (e.g., Stat1, Stat3, Meis1, Pbx1, Klf6, Nfia) (Supplemental Table S1).
A G1-specific depletion in old LT-HSCs
Age did not typically affect the proportion of cells within each cluster, with one notable exception of old LT-HSCs, where the frequency of cells in cluster 1 (G1/S cluster) was significantly decreased in old versus young cells (7.6% vs. 22%; P = 0.0023, hypergeometric test) (Fig. 2E; Supplemental Fig. S3E). The depletion of cells in the G1/S phases was specific to old LT-HSCs and was not observed for either old ST-HSCs or MPPs. We confirmed a decrease in the frequency of old LT-HSCs that are in the G1 phase by an orthogonal approach of staining BM cells with Ki67/Hoechst paired with bromodeoxyuridine (BrdU) incorporation followed by FACS (approximately two- to threefold decrease; P = 0.012 independent samples t-test) (Fig. 2G,H) with similar proliferation rates by in vivo BrdU incorporation (mean 19.6% and 20.1% for old and young, respectively; P = 0.916 independent samples t-test) (Fig. 2I,J).
A decreased proportion of cells observed in specific cell cycle phases (G1/S here) suggests more rapid transitions through these phases (Kafri et al. 2013), implying shorter G1 and/or S phases. Multiple studies, in mouse and human cells, established that G1 length varies widely among different cell types, is short specifically in self-renewing cells, and increases with differentiation (Li et al. 2012; Coronado et al. 2013). We thus reasoned that the same trend may hold in LT-HSCs, where aging could be accompanied by a decrease in G1 length and LT-HSC accumulation.
To refine our hypothesis of a G1-specific depletion in old LT-HSC, we performed a higher-resolution analysis of cell cycle progression using our single-cell profiles. Plotting the average expression of G1/S versus G2/M genes in each cell revealed a complete cell cycle trajectory (Fig. 2F). By using the location of cells on that trajectory, we ranked cells with respect to their cell cycle progression (Kafri et al. 2013; Trapnell et al. 2014). We roughly estimated the range of cell ranks that might correspond to distinct phases of the cell cycle, with ∼20%, 6%, and 9% of cells in G1, S, and G2/M, respectively, and 65% of cells in the G0 noncycling state (Supplemental Fig. S4B; Supplemental Table S2). Comparing the frequencies of old to young LT-HSCs along the cell cycle progression ranks revealed the most pronounced depletion within the late G1 phase (approximately fourfold) (Supplemental Fig. S4C–E). To confirm this finding, we isolated and sequenced an additional 200 single cells of LT-HSCs and ST-HSCs from an additional set of old C57BL/6 mice. Once again, we observed a significant age-dependent decrease in the frequency of cells in G1 phase among LT-HSCs but not ST-HSCs (Supplemental Fig. S4D). Taken together, our experiments and analyses all support the finding that old LT-HSCs specifically have a smaller pool of cells in the G1 phase.
G1 phase is a sensitive period during which cell fate decisions are made (Takahashi et al. 1995; Sherr 2000; Calegari and Huttner 2003; Massague 2004; Pauklin and Vallier 2013). Furthermore, mouse embryonic stem cells continue to self-renew and thus avoid differentiation by eliminating or greatly shortening their early G1 phase (Savatier et al. 1994; White et al. 2005). Recent work has also demonstrated that unlike the majority of slow-cycling cells with stochastic and inefficient reprogramming to pluripotency, a small subset of hematopoietic cells with rapid G1 can be deterministically reprogrammed, raising the possibility that G1 length may be linked to dedifferentiation (Guo et al. 2014). We therefore speculated that a short G1 in old LT-HSCs might be linked to a dedifferentiated state. This hypothesis is also consistent with recent epigenomic analyses of young and old HSCs (Sun et al. 2014).
Opposing transcriptional states for aging and differentiation in HSCs
To test for a possible link between HSC aging and self-renewal/differentiation, we first excluded the 367 cycling cells (to avoid the dominant effects of the cell cycle) and performed a joint PCA on the remaining 692 noncycling cells (contains all cell types and ages). Each of the first two PCs segregated HSCs by cell type and age (Fig. 3A–E; for additional PCs, see Supplemental Fig. S5A). Accordingly, with respect to these two PCs, young LT-HSCs are clearly distinct from the old LT-HSCs (due to an age effect) (Fig. 3B,E, the same plot as in Fig. 3A but only specific pairs of populations that differ by age are displayed), as well as from the young ST-HSCs (due to a differentiation effect) (Fig. 3C,E, the same plot as in Fig. 3A but only specific pairs of populations that differ by differentiation are displayed). Interestingly, young LT-HSCs are similar to the old ST-HSCs that differ in both age and differentiation state (Fig. 3D,E). Consistently, both PC1 and PC2 were inversely associated with differentiation and aging, scoring high for cells both from younger animals (Fig. 3A,B) and from more differentiated cells (e.g., MPPs and ST-HSCs from either young or old mice score higher than their age-matched LT-HSC counterparts) (Fig. 3A,C) and scoring low both for old cells and for less-differentiated cells. These results were reproduced also with the expression profiles from the replicate experiment (about 200 LT-HSCs and about 200 ST-HSCs; different mice obtained, sorted, and profiled months apart) (Supplemental Fig. S5B). Thus, the transcriptional program of old LT-HSCs and ST-HSCs resembles a less differentiated state than their young counterparts, which could reflect loss of balance between self-renewal and differentiation. This could be linked to the recent finding of a developmental switch, where in embryonic development LT-HSCs are the main contributors for most of the blood-cell production in unperturbed hematopoiesis, while in adult life, ST-HSCs assume that role (Busch et al. 2015). To investigate the clonogenic potential of individual LT-HSCs and ST-HSCs from young and old mice regardless of their reconstituting activity in vivo, we assessed colony formation on methylcellulose. While young LT-HSCs and ST-HSCs gave rise to a similar number of colonies, both old counterparts formed significantly more colonies (1.4 and 1.3 increase in LT-HSCs [P < 0.001] and ST-HSCs [P = 0.01], respectively, independent samples t-test) (Fig. 3F).
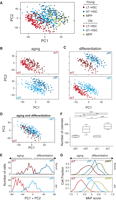
HSC aging and differentiation are associated with opposite expression programs. A joint PCA was performed for all noncycling cells, and each of the top two PCs distinguishes cells by their cell type and age, with higher scores for young and differentiated HSCs and lower scores for old and less-differentiated cells. (A–D) Each plot shows the loadings of PC1 and PC2, colored based on their cell type and age for cells from all six populations (A) or from specific pairs of populations that differ by age (B), differentiation (C), or both (D). (E) Distribution of PC1 + PC2 scores for young (top) and old (bottom) LT-HSCs and ST-HSCs. Aging is associated with a decrease in the PC1 + PC2 scores, and differentiation is associated with an increase in the PC1 + PC2 scores. (F) Colony formation assays using methylcellulose. Two hundred fifty of either young or old LT-HSCs and ST-HSCs were plated on methylcellulose (n = 5). Colonies were counted on day 10. The colony numbers are averages of duplicate measurements of each individual mouse. Statistically significant differences are as follows: (***) P < 0.001, (*) P < 0.05. (G) Distribution of the megakaryocyte progenitor (MkP) signature scores, defined as the average normalized expression of MkP-enriched genes (Sanjuan-Pla et al. 2013) (x-axis) for LT-HSC (red), ST-HSC (blue), and MPP (green) in young (top) and old (bottom) mice.
Notably, PC1 and PC2 also correlate with an expression signature of megakaryocyte progenitors (MkP) (Fig. 3F; for other signatures, see Supplemental Fig. S4C; as defined by Sanjuan-Pla et al. 2013), such that old cells express, on average, higher levels of MkP signature genes than their young counterparts, and less-differentiated cells have higher MkP expression levels than more differentiated cells (P < 10−6 for all pairwise comparisons by Mann–Whitney U test) (Fig. 3F). This is largely consistent with the reported myeloid bias of old HSCs (Rossi et al. 2005), but MkP signature genes, and more generally, myeloid-related genes appear to account for only a minority of a larger expression program that is altered during HSC differentiation and aging. For example, the 792 genes that are enriched in at least one of four myeloid progenitor cell types (MkP, pre-CFU, preMegE, preGM) (Sanjuan-Pla et al. 2013) include only 24 of the top 100 genes that correlate negatively with PC1 + PC2, and therefore increase with age, and only 16 of the top 100 genes that correlate positively with PC1 + PC2.
To identify candidate genes that may underlie this inverse relationship, we compared the expression of each gene between pairs of populations that differed by differentiation (LT-HSCs vs. ST-HSCs) or by age (young vs. old) but were matched for the other parameter. Overall, we performed three comparisons for the effects of differentiation (LT-HSCs vs. ST-HSCs in young mice and in two replicates of old mice) and four comparisons for the effects of aging (young vs. old LT-HSCs and ST-HSCs, each with two replicates) to first identify significant differences (P < 0.05, two-sample t-test; no correction for multiple tests) between each pair of conditions. We then identified 78 genes with a consistent aging and differentiation effect (FDR < 0.05; Methods) (Supplemental Table S3). Of these, all but one gene (Ctnnal1) showed an opposite trend with aging and differentiation, suggesting an inverse relationship between these variables (Fig. 4A,B). This list of genes includes several important regulators of hematopoiesis and/or self-renewal, such as Flt3, Cd34, Vwf, Il16, Nfia, Id2, Itgb3, Runx1t1, and Cd44 (Fig. 4A). Expression changes for some of these genes were consistent with a potential imbalance between self-renewal and differentiation or a defect in the differentiation of old LT-HSCs, as Flt3 and Cd34 (lowest in old LT-HSCs) have previously been linked to HSC differentiation. We further validated at the protein level that CD34 and FLT3 decrease with aging and increase with differentiation (Fig. 4C).
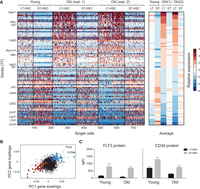
An inverse relationship between the transcriptional signatures of aging and differentiation. (A) A gene signature of aging and differentiation. (Left) Heatmap showing the relative expression levels of 77 genes (rows) significantly associated with aging and differentiation in all noncycling cells of LT-HSCs and ST-HSCs. Cells (columns) are sorted by age and within each age by cell type. Genes above the horizontal black bar are higher in LT-HSCs than in ST-HSCs and in old versus young cells. Genes below the horizontal black bar are higher in ST-HSCs than in LT-HSCs and in young versus old cells. (Right) The average expression of each gene (row) over all the noncycling cells from each combination of cell type and age (column). (B) Genes in the signature have correlated loadings on PC1 and PC2. Shown are the PC1 (x-axis) and PC2 (y-axis) loadings for each gene. Genes in the signature in B are marked in large points, colored in blue and red, respectively, for either high or low loadings for both PCs. (C) CD34 and FLT3 proteins are both decreased with age and increased with differentiation. Shown is the median fluorescence intensity (MFI; y-axis) of fluorescent-conjugated FLT3 (left) and CD34 (right) protein in young and old LT-HSCs (black) and ST-HSCs (gray).
An age-independent expression program among MPPs but not other cell types
While the aging-differentiation expression program described above appears to dominate the variability among all noncycling HSCs (i.e., from the three cell types together), we next asked whether we could identify additional sources of variability within each individual cell type. PCA performed independently on the noncycling LT-HSCs (Fig. 5A) and ST-HSCs (Fig. 5B; as opposed to a joint PCA for all cells and ages in Fig. 3) showed that aging dominates the cell-to-cell variability within both of these cell types, and we could not find additional major independent sources of variability, such as additional subsets of cells. This suggests that most variations discernible within LT-HSCs or ST-HSCs and not related to cell cycle status are associated with cell-intrinsic changes that manifest rather uniformly across each population rather than with the emergence of specific subsets. We discuss how this relates to known functional heterogeneity below (see Discussion).
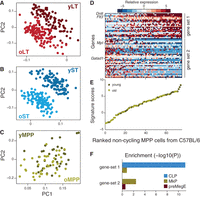
Subsets of cells with lymphoid- and myeloid-like transcriptional bias are discernible within immunophenotypically defined MPPs in C57BL/6 mice. (A–C) MPPs profiles are not distinguishable by age. PCA was performed independently for noncycling cells of each of LT-HSCs (A), ST-HSCs (B), and MPPs (C). Each plot shows the loadings of PC1 and PC2, colored based on cell type and age. Higher scores for young HSCs and lower scores for old cells are characteristic for LT-HSCs (A) and ST-HSCs (B), but not for MPPs (C). (D) Two distinct modules in MPPs. Heatmap shows the expression of genes from two distinct gene sets (rows; gene set 1 indicates lymphoid-biased; gene set 2, myeloid-biased) across all noncycling MPPs (columns). (E) Noncycling MPPs from both young and old mice form a continuous spectrum along the two states. Shown are the signature scores (average normalized expression of gene set 1 minus that of gene set 2) for each noncycling MPP from young (dark green) or old (light green) mouse. MPPs are ranked by increasing scores (x-axis). (F) Gene set enrichment analysis based on defined progenitor sets (CLP, MkP, and preMegE) within gene sets (gene set 1 and gene set 2) defining two subsets of MPPs in C57BL/6.
In contrast, we found that among noncycling MPPs, expression variability is largely independent of age (Fig. 5C), and we detected an age-independent expression program whose levels vary considerably within the immunophenotypically defined MPP compartment (Fig. 5D). This uncovered two cell states within MPPs that have discrete molecular signatures—gene set 1 and gene set 2 (Fig. 5D; Supplemental Table S4)—yet represent part of a continuous spectrum, irrespective of age (Fig. 5E). Enrichment analysis using gene sets from lineage-restricted progenitors (Pronk et al. 2007; Sanjuan-Pla et al. 2013) revealed a lymphoid bias for gene set 1 and premegakaryocyte/erythroid and megakaryocyte progenitor bias for gene set 2 (Fig. 5F). This highlights MPPs as a heterogeneous population likely including both lymphoid-biased and myeloerythroid-biased progenitors, in line with MPPs giving lymphoid and myeloerythroid reconstitution in competitive transplants (Oguro et al. 2013).
Age-associated changes are conserved between mouse strains
Life span and aging vary between mouse strains. For example, C57BL/6 mice are long-lived compared with the short-lived DBA/2 mice (Turturro et al. 1999). To test the generality of our observations, we also examined LT-HSCs, ST-HSCs, and MPPs in young and old mice from the DBA/2 strain, which originates from a distinct breeding lineage (Fox 1997).
By using SLAM markers with conserved expression among mouse strains (Kiel et al. 2005) and ages (Fig. 6A; Supplemental Fig. S6A,B; Yilmaz et al. 2006), we quantified the frequency of LT-HSCs, ST-HSCs, and MPPs in BM by flow cytometry (Fig. 6B) and stem cell compartment (LSK) of young and old DBA/2 mice (Supplemental Fig. S6C). We observed an age-associated increase of LT-HSCs frequency, albeit to a lower extent (approximately twofold, P < 0.001) than in C57BL/6 (approximately sixfold, P < 0.001) (Fig. 6B). Moreover, hemoglobin concentration and red blood cell (RBC) numbers were significantly lower in aged mice (hemoglobin 10.3 vs. 12.7 g/dL, P = 0.001, and RBC 8.2 vs. 9.2 × 106/µL, P = 0.04; independent samples t-test), indicating anemia, than in C56BL/6 mice (hemoglobin 11.8 vs. 14 g/dL, P = 0.001, and RBC 8.38 vs. 9.9 × 106/µL, P < 0.001).
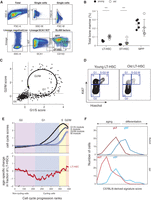
Age associated changes are conserved in DBA/2. (A) Gating strategy used to isolate LT-HSCs (LSK CD150+CD48−), ST-HSCs (LSC CD150−CD48−), and MPPs (LSK CD150−CD48+) from the BM of young (6–12 wk) DBA/2 mice. (B) LT-HSC compartment expands during aging. Shown are frequencies of LT-HSCs, ST-HSCs, and MPPs (x-axis) in young (black) and old (white) DBA/2 mice as a percentage of BM. Statistically significant differences: (***) P < 0.001. (C) Cell cycle trajectory inferred from single-cell RNA-seq. Shown is the average expression of G1/S genes (x-axis) and G2/M genes (y-axis). The arrow and labels reflect inferred cell cycle progression. (D) Representative FACS plots for Ki67/Hoechst intracellular staining in young and old LT-HSCs from DBA/2 mice. (E) Cell cycle distribution changes with age. (Top) Cells were ordered according to their inferred cell cycle progression, and the average expression of G1/S, S, and G2/M genes (y-axis, curves from dark to light gray) was calculated with a sliding window of 11 cells (x-axis). The first approximately 217 cells are “noncycling,” and only a small portion of them is depicted in the graph. The G0/G1 approximate border was defined as the first position with a positive G1/S score (i.e., above the average of all cells), and other borders were approximated by manual inspection of the figure. (Bottom) For each cell type, the log2 of the ratio between percentages of old cells divided by the percentage of young cells along the inferred cell cycle progression (with a sliding window of 100 cells) is shown. Shaded colors reflect the inferred cell cycle phases; cells are ordered by the analysis of the top panel. (F) Distribution of the C57BL/6-derived signature scores for young and old LT-HSCs and ST-HSCs, defined as the average normalized expression of young LT-HSC–enriched (dark red), old LT-HSC–enriched (light red), young ST-HSC–enriched (dark blue), and old ST-HSC–enriched (light blue) genes (x-axis), respectively.
Next, we used two approaches to compare the cell cycle distribution between young and old LT-HSCs from DBA/2 mice. First, we performed single-cell RNA-seq with prospectively isolated LT-HSCs, ST-HSCs, and MPP from young and old DBA/2 mice. Our high-resolution cell cycle analysis again revealed the entire cell cycle trajectory (Fig. 6C) and a modest (approximately twofold) depletion of old LT-HSCs in a region of the cell cycle trajectory that presumably reflects the early G1 phase (Fig. 6E). Second, staining BM with Ki67/Hoechst showed a decrease in the frequency of old LT-HSCs that are in the G1 phase (Fig. 6D; Supplemental Fig. S6D). FACS analysis showed a similar, albeit nonsignificant, trend for ST-HSCs (Supplemental Fig. S6D). Taken together, a specific depletion of old LT-HSCs in the G1 phase appears to be conserved between the C57BL/6 and DBA/2 strains.
Finally, we examined the conservation of the expression programs described above. PCA of all noncycling DBA/2 cells recapitulated the results in C57BL/6 and showed that the first two PCs are negatively associated with age (Supplemental Fig. S6E,F) and positively associated with differentiation (i.e., ST-HSCs vs. LT-HSCs) (Supplemental Fig. S6E,G). As in C57BL/6, young LT-HSCs are more similar to old ST-HSCs than to old LT-HSCs in DBA/2 (Supplemental Fig. S6H). Moreover, while the genes associated with PC1 and PC2 partially differ between the two strains, by using the top genes that discriminate age and differentiation states in C57BL/6 as a signature, we recapitulated the same effect in DBA/2 (Fig. 6F). As in C57BL/6, PCA among noncycling cells within each cell type showed the dominant effect of age for LT-HSCs and ST-HSCs, but not for MPPs (Supplemental Fig. S7A–C). MPPs showed a similar pattern as in C57BL/6, where two cell states—one lymphoid-biased and the other myeloid-biased (Supplemental Fig. S7G)—coexist within the phenotypically defined MPPs (Supplemental Fig. S7D,E). Taken together, all of our main results in C57BL/6 are largely recapitulated in DBA/2, further strengthening their generality.
Discussion
Cellular heterogeneity of young HSCs
Despite markers that can give high levels of HSC purity, HSC populations remain functionally heterogeneous both with respect to their self-renewal potential upon transplantation (Benveniste et al. 2010; Morita et al. 2010) and in the ratio of myeloid to lymphoid cells that they generate upon transplantation into irradiated mice (Muller-Sieburg et al. 2002; Dykstra et al. 2007; Kent et al. 2009; Beerman et al. 2010; Challen et al. 2010; Morita et al. 2010). Here, we examined if this retrospective heterogeneity would be prospectively reflected in the transcriptional profiles of single cells. To approach this question, we profiled, for the first time, a very large number of extremely rare single cells from three different cell types (LT-HSCs, ST-HSCs, and MPPs), two ages (young and old), and two strains (C57BL/6 and DBA/2) using single-cell RNA-seq. This allowed us to assess the current or prospective view of potential HSC attributes through a direct approach of surveying the current transcriptional state of individual cells in steady-state unperturbed hematopoiesis to complement retrospective analysis by transplantation (perturbed hematopoiesis).
We identified extensive transcriptome variability among HSCs and associated it to the cell cycle, differentiation, and age. After accounting for these sources of variability, we do not observe discrete subsets of cells within LT-HSCs and ST-HSCs from either age or strain of mice. We relied on a SLAM markers sorting strategy, one of the best strategies to enhance HSC purity, as 47% of single LSK CD150+CD48− BM cells (one in 2.1) give long-term multilineage reconstitution (Kiel et al. 2005). Similarly, previously observed patterns of long-term repopulation (α, β, δ, γ), although sorted using a different strategy (isolated from CD45midlin−Rho−SP), occur in high frequencies: 27%, 39%, 22%, and 12% for α, β, δ, and γ cells, respectively (Dykstra et al. 2007). These proportions are sufficiently high to ensure that if these 47% of LT-HSC SLAM cells or α, β, δ, and γ cells have a transcriptional profile or signature that prospectively (prior to transplantation) distinguishes them from other cells, they will be readily discernible in the approximately 600 LT-HSCs we have analyzed. Surprisingly, that has not been the case. While we have not seen a discernible profile that characterizes a distinct subset of LT-HSCs (or ST-HSCs), our computational methods readily discern subpopulations within MPPs (which may indicate an initial specification of HSCs into myeloerythroid and lymphoid lineages); thus the lack of detectable HSC transcriptional subpopulations, within a given cell type and age, is likely not due to a simple weakness in computational methods. In particular, we extensively searched for recently reported platelet-biased LT-HSCs within our single-cell data set using previously defined signatures based on population studies (Sanjuan-Pla et al. 2013), but we have not seen any subpopulations of cells in either young and old mice from either strain (Supplemental Fig. S8A–D).
The absence of discernible subpopulations in our study does not in any way preclude functional heterogeneity, but it suggests that this heterogeneity, for example, may be “stochastic” (Till et al. 1964; Abkowitz et al. 1995; Kirkland 2004; Roeder et al. 2005) or related to a very small number of key genes (rather than a broad state) or may manifest only under specific signals, such as those the transplanted cells may encounter upon transplantation (Trentin 1971; Metcalf 1998; Moore and Lemischka 2006). Another possibility is that the cell fate choice is coupled inextricably to cell cycle states, in which we have observed clear changes with age.
Compositional and coordinated expression changes associated with HSC aging
Aging could, in principle, be associated with changes in the frequency of certain phenotypes or expression programs (clonal selection model) or may be driven by the concerted change of cell intrinsic states across the entire population of old HSCs (global population shift model). Our results show evidence for both compositional changes and coordinated, population-wide shifts in cell states, demonstrating the power of single-cell analysis in distinguishing these models. We observed, upon aging, a conserved reduction specifically of LT-HSCs in G1 phase, reflecting a change in proportion of that subpopulation of cells. Previous studies reported inconsistent cell cycle changes ranging from increased to unchanged to decreased proliferation in old LT-HSCs (Flach et al. 2014). In our hands, cell cycle analyses consistently showed a decreased number of G1 cells in two mouse strains (C57BL/6 and DBA/2) and by two independent approaches: as inferred from single-cell RNA-seq data (in duplicate experiments done weeks apart, and based on the entire gene signature) and by Ki67/Hoechst staining paired with BrdU. Interestingly, a recent study reported decreased expression of genes related to cell cycle, DNA replication, and DNA base excision repair (which are normally up-regulated during G1/S) in old LT-HSCs (Sun et al. 2014). Our results suggest that this observation may not be due to down-regulation of those genes across most old LT-HSCs but rather due to a decreased proportion of the subset of G1/S cells that highly express those genes. Moreover, our single-cell RNA-seq approach revealed an entire cell cycle trajectory based on coexpression of a large number of genes that is consistent between cell types and species (Supplemental Fig. S2). We detected amid quiescent cells, expression programs that are observed in practically every single cell we sampled from each cell type, age, and strain without identifiable discrete cell subsets within LT-HSCs and ST-HSCs.
Self-renewal vs. differentiation-biased expression programs in HSCs
Our analysis revealed an expression program of old HSCs that differs from young HSCs and is diametrically opposed to the expression program associated with differentiation (Fig. 3). In particular, old ST-HSCs resembled transcriptionally young LT-HSCs, which may be related to the recent demonstration that adult ST-HSCs nearly fully self-renew and serve as the main source of hematopoietic maintenance in mice (Busch et al. 2015). This raises the possibility that HSCs can occupy distinct positions in gene expression space such that those that are closer to the state of differentiated cells have a higher propensity for differentiation (differentiation biased), while those that are located in the opposite direction have a higher propensity for self-renewal (self-renewal biased). Accordingly, aging could shift LT-HSCs toward the self-renewal–biased state and away from the differentiation-biased state (Fig. 7). This is consistent with a recently suggested link between aging and self-renewal (Sun et al. 2014). Taken together, age-associated accumulation of LT-HSCs and their functional decline may be a consequence of the imbalance between self-renewal and differentiation in favor of the former, and this imbalance may be reflected by a self-renewal–biased expression program. While these changes have consequences for the early steps of hematopoiesis, it remains to be determined what effects these may cause to the entire hematopoiesis (e.g., clonal hematopoiesis) (Holstege et al. 2014). Additional studies would be required to test this hypothesis and examine the functional implications of a decrease in the differentiation-related expression signature and whether it reflects increased self-renewal capacity. This may require developments of new tools given that ST-HSCs are the main contributors to steady-state hematopoietic production in unperturbed conditions, while they are relatively short lived in standard transplantation experiments (Busch et al. 2015).
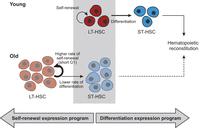
A model of age-dependent changes in LT-HSCs. Young LT-HSCs (red; top) maintain an appropriate balance between efficient self-renewal (semi-circular arrow) and differentiation into ST-HSCs (horizontal arrow) that then further differentiate to reconstitute hematopoiesis. In contrast, old LT-HSCs (bottom) are inappropriately shifted toward self-renewal (thick semi-circular arrow) and thereby an accumulation of LT-HSCs (depicted by more copies of old LT-HSCs), ST-HSCs reduction (depicted by less copies of ST-HSCs), and less efficient reconstitution of hematopoiesis (dashed arrow). This can be due to either a short G1, which limits the capacity of old LT-HSCs to receive differentiation signals, or an expression program that resembles a less differentiated state and might reflect defects in differentiation, or both, as these might be causally linked.
Our analysis identified a set of genes whose expression is both aging and differentiation dependent (Supplemental Table S3), and these include several potentially causal regulators of HSC self-renewal and differentiation. Flt3 and Cd34 have been implicated in HSC differentiation (Osawa et al. 1996; Adolfsson et al. 2001; Yang et al. 2005; Buza-Vidas et al. 2011) and are expressed at lower levels in old versus young LT-HSCs. Notably, FLT3 is a cytokine tyrosine kinase receptor, which is mutated in about a third of acute myeloid leukemia patients (Small 2006). Conversely, Nfia, Id2, and Itgb3 have been implicated in self-renewal (Miller et al. 2013; Imayoshi and Kageyama 2014; van Galen et al. 2014) and are expressed at higher levels in old versus young LT-HSC. Dedicated studies are required to characterize these putative candidates in this context.
A cell cycle–dependent interplay between aging and differentiation
We observed a depletion in cells in the late G1 phase in old LT-HSCs, suggesting a faster transition through G1 and into S, which may shed light on the reduced expression of cell cycle–related genes that was previously observed in old HSCs at the population level (Chambers et al. 2007; Sun et al. 2014). This is reminiscent of the short G1 phase in mouse and human embryonic stem cells. In embryonic stem cells, this is achieved by avoiding a normal G1 checkpoint, which shifts the balance from differentiation to self-renewal (Savatier et al. 1994; White et al. 2005). Old LT-HSCs might avoid the G1 checkpoint through the same or distinct mechanisms. Interestingly, one of the genes whose expression increases with age and decreases with differentiation (thereby is maximally expressed in old LT-HSCs) was Polo-like kinase 2 (PLK2), a known checkpoint regulator that controls progression during G1 and early S phases (for review, see van de Weerdt and Medema 2006). PLK2 inactivation was shown to extend the length of the cell cycle and delayed the entry into S phase (Ma et al. 2003), suggesting that PLK2 up-regulation in old LT-HSCs could facilitate rapid progression through G1.
Since asymmetric distribution of cell-fate determinants enables two daughter cells to follow different fates, the cell cycle may be linked to cell fate through the establishment of cell polarity, as shown in Caenorhabditis elegans and Drosophila melanogaster (Noatynska et al. 2013). Previous work has shown that young LT-HSCs undergo both symmetric and asymmetric division and that the precise balance between the two is affected by various signals (Wu et al. 2007). Furthermore, LT-HSCs become largely apolar with age (Florian et al. 2012), raising the possibility that the aging-related changes described here are linked to the loss of cell polarity. Accordingly, it is possible that apolar old LT-HSCs, which traverse through G1 phase faster than their younger counterparts, undergo preferential symmetric divisions, thereby leading to amassing of LT-HSCs with age.
Taken together, it is tempting to speculate that the accumulation of LT-HSCs, their short G1 phase, and self-renewal-biased expression program are mechanistically coupled. This could be accomplished either by an altered cell cycle progression and a shorter G1 phase driving a self-renewal-biased expression program or by an expression program affecting the cell cycle, which, in turn, results in a shorter G1 phase. Either scenario may ultimately lead (perhaps through loss of polarity) to a symmetric cell division, where both daughter cells maintain LT-HSC identity and, thereby, drive the increase of LT-HSCs frequencies in BM and at the same time a disproportionally smaller number of differentiated cells (i.e., ST-HSCs in the LSK compartment). Additional work will be required to further examine these possibilities and the role of potential candidate genes such as PLK2 in the connection among cell cycle, aging, and differentiation-related expression states.
Methods
Isolation of hematopoietic stem and progenitor cells
Young (2- to 3-mo-old) female C57BL/6 and DBA/2 mice were purchased from Taconic and The Jackson Laboratory, respectively. Old female C57BL/6 (>22-mo-old) and DBA/2 (20-mo-old) mice were obtained from the National Institute of Aging Mice and housed in the Boston Children's Hospital Animal facility (IACUC 1012-104-15).
For single-cell sorting (BD FACSAriaII), total BM cells were isolated from long bones and underwent erythrocyte lysis and CD117 enrichment prior to antibody staining (CD11b, Gr1, CD45R, CD3e, TER119, CD117, Sca1, CD48, CD150; all from eBioscience and BioLegend). LT-HSCs, ST-HSCs, and MPPs were double-sorted into 96-well plates preloaded with 5 µL of lysis buffer (TCL lysis buffer, Qiagen) supplemented with 1% (v/v) 2-mercaptoethanol (Qiagen) and flash frozen. For populations, cells were sorted directly into lysis buffer and RNA extracted using PrepEase (Affymetrix).
For cell cycle, we used BD Cytofix/Cytoperm and the Ki-67 kit (BD) following the manufacturer's instructions.
For in vivo BrdU incorporation, mice were intraperitoneally injected with two doses of BrdU (BD Pharmingen), 8 and 2 h before sacrifice, at 50 μg/g b.wt., and we used the BrdU flow kit (BD Pharmingen) for detection.
For colony-formation assays, 250 FACS-sorted LT-HSCs or ST-HSCs from young and old C57BL/6 mice were plated on MethoCult (M3434, STEMCELL Technologies) according to the manufacturer's instructions. The colonies were counted on day 10.
RNA-seq library preparation and initial data processing
We prepared and profiled libraries as previously described (Patel et al. 2014) from 1152 individual cells. Sequencing data were processed as previously described (Patel et al. 2014). Before all subsequent analyses, we filtered and centered the data. First, we filtered out cells with less than 2500 genes with log2(TPM + 1) > 2. Second, we excluded genes whose log2(TPM + 1) < 4 in the aggregated data for each of the six populations. Third, we centered the data by subtracting for each gene its average expression [log2(TPM + 1)] across all cells. MATLAB's PCA function was used with default parameters.
Clustering based analysis of cell cycle state
Cell cycle genes were defined as those with a “cell cycle process” Gene Ontology annotation (downloaded from MSigDB version 3.1) (The Reference Genome Group of the Gene Ontology Consortium 2009) or identified as cycling in HeLa cells (Whitfield et al. 2002). We defined four cell cycle signatures (G1/S, S, G2/M, and M) as the average expression [log2(TPM + 1)] of phase-specific subsets of the cell cycle genes as defined previously in synchronized HeLa cells (Whitfield et al. 2002). We refined these signatures by averaging only over those genes whose expression pattern in our data correlated highly (r > 0.5) with the average signature of the respective cell cycle phase (before excluding any gene) in order to remove the influence of genes that were previously detected in HeLa cells but do not appear to cycle in our data.
High-resolution analysis of cell cycle progression
Plotting the average of the G1/S and S signatures versus the average of the G2/M and M signatures for each cell (Fig. 2D; Supplemental Fig. S3A) showed an approximate circle, which we assume reflects all phases of the cell cycle. Based on this assumption, we ordered all cells according to the apparent progression along the cell cycle and assigned a rank of cell cycle progression to each cell (Supplemental Fig. S3A; Supplemental Table S1).
Identification of aging- and differentiation-dependent differentially expressed genes
Two-sample t-tests with a P-value of 0.05 and no correction for multiple testing were initially used to identify potentially significant differential expression in comparison of each pair of populations with matched cell types but different age (age effects) or matched age but different cell type (differentiation effects).
Data access
Sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE59114.
Acknowledgments
We thank Alex Shalek, Rahul Satija, Orit Rozenblatt-Rosen, and Dawn Thompson for scientific discussions; Candace Guiducci for project management; Leslie Gaffney for assistance with artwork; and the Broad Genomics Platform for all sequencing work. We thank DFCI and Joslin/HSCI Flow Cytometry Cores for excellent flow cytometry support. This work was supported by an EHA Research Fellowship award from the European Hematology Association (M.S.K.), EMBO Long Term Fellowship (M.S.K.), Foundation for Polish Science (M.S.K.), HFSP Long Term Fellowship (I.T.), Rothschild Fellowship (I.T.), German Cancer Foundation (D.H.), Leukemia and Lymphoma Society Scholar Award (B.L.E.), National Institutes of Health (NIH) (P01 CA066996, B.L.E.), HHMI (A.R.), NIH CEGS Award (1P50HG006193-01, A.R.), Koch Institute Support (core) Grant from the National Cancer Institute (P30-CA14051, A.R.), and the Klarman Family Foundation (A.R.). A.J.W. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.192237.115.
- Received March 19, 2015.
- Accepted September 30, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








