
Differential retention and divergent resolution of duplicate genes following whole-genome duplication
- 1Department of Biology, Indiana University, Bloomington, Indiana 47408, USA;
- 2National Center for Genome Analysis Support at Indiana University, Bloomington, Indiana 47408, USA
- Corresponding author: milynch{at}indiana.edu
-
↵3 These authors contributed equally to this work.
Abstract
The Paramecium aurelia complex is a group of 15 species that share at least three past whole-genome duplications (WGDs). The macronuclear genome sequences of P. biaurelia and P. sexaurelia are presented and compared to the published sequence of P. tetraurelia. Levels of duplicate-gene retention from the recent WGD differ by >10% across species, with P. sexaurelia losing significantly more genes than P. biaurelia or P. tetraurelia. In addition, historically high rates of gene conversion have homogenized WGD paralogs, probably extending the paralogs’ lifetimes. The probability of duplicate retention is positively correlated with GC content and expression level; ribosomal proteins, transcription factors, and intracellular signaling proteins are overrepresented among maintained duplicates. Finally, multiple sources of evidence indicate that P. sexaurelia diverged from the two other lineages immediately following, or perhaps concurrent with, the recent WGD, with approximately half of gene losses between P. tetraurelia and P. sexaurelia representing divergent gene resolutions (i.e., silencing of alternative paralogs), as expected for random duplicate loss between these species. Additionally, though P. biaurelia and P. tetraurelia diverged from each other much later, there are still more than 100 cases of divergent resolution between these two species. Taken together, these results indicate that divergent resolution of duplicate genes between lineages acts to reinforce reproductive isolation between species in the Paramecium aurelia complex.
Whole-genome duplications (WGDs) are widespread among eukaryotic lineages and have been identified in the ancestry of many model systems, including Saccharomyces cerevisiae (Wolfe and Shields 1997), Xenopus laevis (Morin et al. 2006), Danio rerio (Postlethwait et al. 2000), and Arabidopsis thaliana (Simillion et al. 2002). There is increasing support for the hypothesis that two WGDs preceded the radiation of the vertebrate lineage (Panopoulou and Poustka 2005; Hughes and Liberles 2008; Putnam et al. 2008; Decatur et al. 2013), and nearly all land-plant genomes appear to have experienced at least one WGD, with a proposed WGD in the ancestor of all seed plants and another in the ancestor of all angiosperms (Jiao et al. 2011; Amborella Genome Project 2013). Despite their prevalence, the evolutionary ramifications of WGDs remain poorly understood. The WGD in the yeast lineage is one of the best studied; however, only ∼8%–14% of duplicates remain from this event (Wolfe and Shields 1997; Scannell et al. 2007), leaving relatively few genes upon which to draw inferences.
One of the obvious impacts of WGDs is the simultaneous creation of thousands of duplicated genes, which have long been thought to be the major source of raw material for new gene functions (Ohno 1970). Neofunctionalization, whereby one copy acquires a novel beneficial function at the expense of an ancestral function, and subfunctionalization, whereby complementary mutations lead to the partitioning of independently mutable ancestral subfunctions, both lead to long-term preservation of paralogs (Hughes 1994; Force et al. 1999; Lynch et al. 2001; Taylor and Raes 2004; Hahn 2009; Innan and Kondrashov 2010; McGrath and Lynch 2012).
Duplicated genes can also be retained without change of functions. For example, selection for dosage balance between gene products is known to be an important force opposing gene loss after a WGD (Veitia 2002; Papp et al. 2003; Birchler et al. 2005; Veitia et al. 2008).
A number of other gene features such as expression level (Aury et al. 2006; Conant and Wolfe 2008; Gout et al. 2010), essentiality (Conant and Wolfe 2008), protein length and number of domains (He and Zhang 2005), evolutionary rate (Chapman et al. 2006), location in a protein interaction network (Wu and Qi 2010), and number of phosphorylation sites (Amoutzias et al. 2010) also correlate with retention in various organisms. However, because many of these features are expected to be correlated with each other, it is possible that most of these correlations are actually caused by a single gene feature.
Another important possible consequence of WGDs is the emergence of reproductive isolation between subpopulations due to reciprocal gene losses. Reciprocal gene loss (also referred to as divergent resolution) occurs when two subpopulations each lose a different copy in a pair of genes. When individuals from such subpopulations produce hybrid offspring, one-quarter of the F1 gametes and one-sixteenth of the F2 zygotes lack the gene completely (Oka 1988; Werth and Windham 1991; Lynch and Conery 2000; Lynch and Force 2000). If gene loss rates are high enough in both populations, this process will rapidly lead to complete reproductive isolation. In addition, if population subdivision and divergent resolution of duplicates continues over time, speciation events can continue to occur, leading to a nested species radiation. Divergent resolution of single-gene duplicates has been implicated in reproductive isolation in Drosophila (Masly et al. 2006), Arabidopsis (Bikard et al. 2009), and rice (Mizuta et al. 2010; Yamagata et al. 2010). The speciation events in yeast have also been hypothesized to be the result of divergent resolution of duplicates following a WGD (Scannell et al. 2006).
Here, we take advantage of the exceptional characteristics of the Paramecium lineage to study the pattern of gene retention following a WGD and its consequences on species divergence. The initial sequencing and analysis of the P. tetraurelia genome revealed a history of three successive WGDs and provided useful information regarding the evolutionary mechanisms responsible for duplicate-gene retention (Aury et al. 2006; Gout et al. 2009, 2010). Interestingly, P. tetraurelia belongs to a complex of 15 species so similar morphologically and ecologically that they were originally believed to represent only one species (Sonneborn 1975), and it has been postulated that the most recent WGD in the Paramecium lineage precipitated the emergence of the aurelia complex (Aury et al. 2006). We now report the sequence and analysis of two additional members of the complex: P. biaurelia and P. sexaurelia.
Results
The macronuclear genome sequences of P. biaurelia and P. sexaurelia
The choice of these two species was motivated by their different evolutionary distances from P. tetraurelia (Catania et al. 2009), with P. sexaurelia being one of the earliest diverging aurelias following the most recent WGD and P. biaurelia diverging from P. tetraurelia much later (Fig. 1).
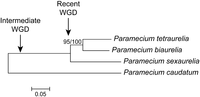
Maximum-likelihood phylogeny of P. biaurelia, P. tetraurelia, and P. sexaurelia, based on the protein sequence of genes predicted in this study (see Methods). P. caudatum was added as a preduplication outgroup (data for P. caudatum comes from McGrath et al. 2014). Approximate locations of recent and intermediate WGDs are indicated. Bootstrap value out of 100 replicates for the P. biaurelia–P. tetraurelia node is indicated. Scale bar represents 0.05 amino acid substitutions per site.
Sequencing of the P. sexaurelia macronuclear genome resulted in an assembly of ∼68 Mb (including estimated gap sizes) composed of 547 total scaffolds, 230 of which are longer than 2 kb and 179 of which are longer than 50 kb (Supplemental Table 1). The P. biaurelia genome is slightly larger at 77 Mb, but the assembly is more fragmented. It is composed of 2362 scaffolds, 1426 of which are longer than 2 kb and 408 of which are longer than 50 kb. These are close to the 72-Mb assembly of the previously published P. tetraurelia genome (Aury et al. 2006). The larger number of P. biaurelia scaffolds likely represents sequencing and assembly difficulties and does not reflect any actual differences in chromosome number/length between species, as there are similar numbers of scaffolds ending in telomere sequences in both the P. biaurelia and P. sexaurelia assemblies (Supplemental File 1).
In addition to having a smaller genome size than P. tetraurelia or P. biaurelia, P. sexaurelia also contains fewer annotated genes: 34,939 compared to 39,521 in P. tetraurelia (annotation v1.85) and 39,242 in P. biaurelia (Table 1). Despite the more fragmented P. biaurelia assembly, the fact that we were able to align most genomic segments between P. tetraurelia and P. biaurelia (see below) indicates that we have probably captured and annotated most of the genes in P. biaurelia. PANTHER (Mi et al. 2012) functional predictions are available for 19,305 (49.2%) P. biaurelia genes, 19,072 (48.3%) P. tetraurelia genes, and 17,084 (48.9%) P. sexaurelia genes (Supplemental File 2). The genome assemblies and annotations of P. biaurelia, P. tetraurelia, and P. sexaurelia used in this analysis are available in ParameciumDB (http://paramecium.cgm.cnrs-gif.fr; Arnaiz and Sperling 2011).
Genome statistics for the three P. aurelia species
The overall GC content for the P. tetraurelia genome (28.0%) is substantially higher than that of P. biaurelia (25.8%) and P. sexaurelia (24.1%) (Table 1). The fact that the difference in GC content is stronger in noncoding regions (Supplemental Table 2) suggests that it is due to nonadaptative processes, such as changes in the mutational spectrum or changes in the strength of GC-biased gene conversion (Galtier et al. 2001; Duret and Galtier 2009).
We found that all large scaffolds in P. biaurelia and P. sexaurelia are composed of highly syntenic, paralogous regions (Methods; Supplemental Files 3–5), similar to what was observed in P. tetraurelia (Aury et al. 2006). This confirmed that P. biaurelia and P. sexaurelia share the most recent WGD with P. tetraurelia, and therefore that this most recent WGD predates the aurelia radiation. The paralogous blocks encompass 34,952 tetraurelia genes (88.4% of all genes), 30,804 sexaurelia genes (88.2%), and 29,723 biaurelia genes (only 75.7%, as the more fragmented biaurelia assembly makes annotation of paralogous blocks more difficult).
We also identified tandem duplicates within each genome and found very few (346 in P. biaurelia, 351 in P. tetraurelia, and 280 in P. sexaurelia) (Supplemental File 6). Interestingly, only 3%–12% of identified tandem duplicates are lineage-specific, with the remainder having at least one existing ortholog or WGD paralog (Supplemental File 6). Thus, tandem (single-gene) duplications are extraordinarily rare in the Paramecium lineage.
The level of duplicated gene retention differs among aurelia species
Alignment of paralogous blocks within each genome reveals substantial variation in post-WGD gene retention among the three aurelia species, with P. biaurelia, P. tetraurelia, and P. sexaurelia having retained, respectively, 52.4%, 49.6%, and 41.6% of duplicated genes (P < 10−8 for all two-by-two comparisons, χ2 test). The similar levels of retention between P. biaurelia and P. tetraurelia are consistent with greater shared evolutionary history between these two species (Fig. 1). To ensure that these patterns do not reflect biases arising from differences in assembly quality, we repeated our analysis after removing regions surrounding gaps (Methods). Although doing so slightly increased the retention rate (by ∼2%) for all three species, it had no substantive effect on the between-species patterns (Supplemental Table 3; Supplemental Files 7–9).
Alignment of orthologous and paralogous regions across all three species (Fig. 2; Supplemental File 10) allowed us to detect cases where both gene copies were lost in a species after the most recent WGD (double losses), while at least one other species retained one or both copies. By doing so, we found the duplicate retention rate to be 44.3% for P. biaurelia, 44.9% for P. tetraurelia, and 35.5% for P. sexaurelia (P < 10−7 for all two-by-two comparisons, χ2 test). Although these lower retention rates could point to frequent double losses, it is also possible that they are upwardly biased by species-specific gene gains. Indeed, a majority (80%–90%) of double losses correspond to cases where a gene is annotated in only one copy in one species (solo genes), a possible consequence of lineage-specific gene gains or annotation artifacts rather than actual double losses. The three species differ slightly in the number of solo genes (916 in P. biaurelia, 1158 in P. sexaurelia, and 1504 in P. tetraurelia within the analyzed regions). Removal of these cases of solo genes from the analysis led to increased retention rates, although the pattern among the three species remains essentially the same, with P. biaurelia and P. tetraurelia sharing similar retention rates (57.3% and 58.2%, respectively, P = 0.18, χ2 test), and P. sexaurelia still having the lowest retention rate (46.8%, P < 10−16 for both comparisons, χ2 test). These differences indicate that the evolutionary mechanisms capable of promoting post-WGD gene retention vary in their strength between species.

Example alignment of paralogous and orthologous scaffolds across the three aurelia species. Scaffolds designated with the same letter (A or B) are orthologous to each other. Homologous genes are displayed in matching colors, and genes that have been lost are in white with gray outlines. Sizes of intergenic regions are not to scale in order to show where gene losses have occurred. The orange dot denotes the location of the recent WGD. Regions shown are P. biaurelia A (scaffold_0033:228908–238905), P. biaurelia B (scaffold_0138:105615–111775), P. tetraurelia A (scaffold51_103:147615–158919), P. tetraurelia B (scaffold51_148:107916–119220), P. sexaurelia A (scaffold_142:121990–127294, reverse complemented), and P. sexaurelia B (scaffold_131:113584–123996).
Gene loss rate declines over evolutionary time
Because P. sexaurelia diverged from the P. tetraurelia–P. biaurelia lineage shortly after the most recent WGD (Fig. 1), gene losses shared among all three species likely occurred early after the WGD, while species-specific gene losses correspond to more recent events. For every ancestral preduplication gene, each of the three species can retain 0, 1 of either copy, or both copies, leading to (43 – 1) = 63 possible configurations of gene retention/loss (the case where all three species have lost both copies of a gene is excluded) (Supplemental Fig. 1). We used parsimony to assign gene losses to individual branches of the tree after removing all solo genes (Fig. 3A) and relied on the median dS estimated in this study (see below) to estimate the time since the different speciation events. Though there are few data points, gene retention appears to drop off dramatically within a short period of time after the WGD and then to follow a roughly linear decline (Fig. 3B). A similar pattern has been observed in yeast, where the percentage of retained duplicates follows a power-law curve and then levels out (Scannell et al. 2006).
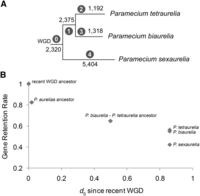
Gene losses and cumulative retention rates across the aurelia phylogeny, based on 13,408 non-solo gene families whose history can be traced from the recent WGD to extant species. (A) Phylogeny based on Figure 1. Positions of losses estimated using parsimony. Black numbers indicate the number of genes lost along each branch; numbers in circles indicate the branch labels referred to in the text. (B) Cumulative gene retention rates over time. Retention levels for extant taxa and inferred retention levels for ancestors based on the tree in A. dS since the WGD is estimated using median dS values between pairs of orthologs and paralogs (see Methods). Note that on the subset of genes used in this analysis, the retention rates for P. tetraurelia and P. biaurelia are not significantly different (0.55 vs. 0.56, respectively, P = 0.12, χ2 test).
Decreasing rate of divergent resolution over time
When both members of a pair of species have lost one copy of a gene, the remaining genes can represent either 1-to-1 orthologs (termed “ancestral/parallel resolution” because either one copy was lost in the ancestor of the two lineages or each lineage lost the same copy in parallel) or 1-to-1 paralogs (termed “divergent gene resolution” because each lineage lost a different copy independently). Because divergent resolution of duplicated genes can lead to reproductive isolation and speciation (Lynch and Conery 2000; Lynch and Force 2000; Shpak 2005) and because WGDs provide thousands of duplicated genes, it has been proposed that the recent WGD in Paramecium promoted the emergence of the aurelia complex. With three aurelia genomes available, we can now quantify the amount of divergent resolution that happened since the recent WGD. We found 2312 cases of divergent resolution and 2741 cases of ancestral/parallel resolution between P. tetraurelia and P. sexaurelia, which represents a 1.2:1 ratio of ancestral/parallel resolution to divergent resolution (Fig. 4A). Although the excess of ancestral resolution is significant (P < 10−9, χ2 test), it is not strong and could be biased by our method of assigning orthology and paralogy (see Methods). In contrast, P. tetraurelia and P. biaurelia share 5821 ancestral/parallel resolutions and only 113 divergent resolutions, a 52:1 ratio (Fig. 4B). Because 80% of the gene losses in P. biaurelia and P. tetraurelia happened before the speciation between these two species (Fig. 3), we conclude that only 1164 (5281 × 0.8) gene losses are parallel, the remaining 4657 being ancestral. This represents a 10:1 ratio of parallel to divergent resolutions, indicating that ongoing gene losses are biased toward parallel resolutions and that the amount of divergent gene resolutions slows down with time. The same pattern has been observed in yeast, and it has been proposed that the stochastic accumulation of mutational divergence between paralogs leads to different subsequent probabilities of retention for each copy (Scannell et al. 2006). In other words, increasing divergence between two paralogs increases the predetermination of the fate of the two copies.
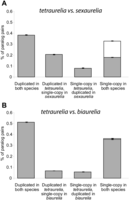
Pairwise comparison of orthologous and paralogous genes between (A) P. tetraurelia and P. sexaurelia and (B) P. tetraurelia and P. biaurelia. The proportion of ancestral genes that are still duplicated in both species, duplicated in one species but single-copy in the other, and single-copy in both species is shown. For genes that are now single-copy in both species, the proportion of divergent resolutions is shown on top (white) and the proportion of ancestral/parallel resolutions is shown below (gray).
Functional analysis confirms the over- and underretention of certain classes of genes
We used PANTHER functional predictions to assess whether some categories of genes were over- or underretained following the WGD. Given that the lineage leading to P. biaurelia and P. tetraurelia diverged from P. sexaurelia very shortly after the WGD (see below), overlap in over- and underretention of functional categories between these two lineages cannot be explained by shared evolutionary history and should reflect the action of convergent evolutionary forces acting to retain paralogs. We observed very similar patterns of gene retention for the different functional categories in all three species (Table 2; Supplemental File 11), indicating that similar evolutionary forces act on the post-WGD gene retention in all three species.
Significantly over- and underrepresented GO terms among retained duplicates
GC content and expression level are correlated with gene retention
In addition to functional category, we asked what other variables might have an effect on duplicate-gene retention, specifically expression level (as measured by mRNA abundance estimated from RNA-seq data) and GC content. After separating aurelia genes into bins based on their GC content, a weighted least-squares regression revealed a significant positive correlation with the duplicate retention rate for all three species (P. biaurelia R2 = 0.94; P. tetraurelia R2 = 0.93; P. sexaurelia R2 = 0.96; P < 10−4 in each case) (Fig. 5A–C). We similarly divided genes into bins based on their expression levels and calculated the retention level of recent WGD duplicates for each bin (Fig. 5D–F). As in the case of GC content, duplicate retention after the recent WGD is positively correlated with the expression level of genes (P. biaurelia R2 = 0.88; P. tetraurelia R2 = 0.69; P. sexaurelia R2 = 0.90; P < 10−4 in each case). In a multiple logistic regression analysis, both expression level and GC content remained significant for all three species (P < 10−3 in each case). For every increase in the GC content of a gene by 0.01, the odds of being retained increases by 6%–10%, while for every increase in the log expression level of a gene by 1, the odds of being retained increase by 5%–11% (Supplemental Table 4). Interestingly, the positive correlation with GC remains strong for genes in different bins of expression level (Supplemental Fig. 5).
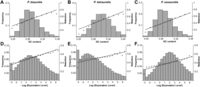
Relationships between duplicate retention and GC content or expression level. GC content (A–C) and log expression level (D–F) are divided into bins. Graphs for P. biaurelia are shown in panels A and D, P. tetraurelia in panels B and E, and P. sexaurelia in panels C and F. For each species, the frequency distribution of the number of genes that fall into each GC content or expression level bin is shown (gray bars), along with the fraction of genes within each bin that are part of a duplicate pair (black dots). Weighted least-squares regression lines of retention on GC content or expression level are shown (black lines).
The correlation between expression level and retention is consistent with a previously proposed model of retention for increased dosage (Gout et al. 2009, 2010). The reason for the relationship between GC content and retention, however, is less clear. One possible explanation is that high GC content reflects strong selective pressure. Indeed, the mutation spectrum of P. tetraurelia is strongly biased toward AT (Sung et al. 2012), as in most other species (Lynch 2010), so that purifying selection appears to be essential for maintaining GC-rich genes. Thus, assuming that genes whose coding sequence is under strong selection are also under selective pressure for post-WGD retention could explain the observed correlation between GC content and probability of retention.
Evidence for widespread gene conversion between paralogs derived from the most recent WGD
Gene conversion between WGD duplicates can lead to an extended maintenance of paralogs (Walsh 1987; Teshima and Innan 2004; Takuno et al. 2008). By homogenizing the sequence of paralogs, frequent gene conversion slows down divergence between paralogs, encouraging joint retention or loss of both copies. Therefore, assuming that loss of both copies is strongly deleterious, frequent gene conversion will promote retention of paralogs. The lower sequence divergence between intraspecific paralogs relative to that between interspecific paralogs—expected given gene conversion—can be detected by contrasting the patterns of synonymous divergence (dS). For all three species, the median dS of intraspecific paralogs was significantly lower than that of interspecific paralogs (Table 3; Fig. 6; Supplemental Fig. 2), suggesting that gene conversion has been operating to homogenize paralogous sequences. Because gene conversion events that occurred before the speciation between the P. biaurelia and P. sexaurelia lineages cannot be detected by this analysis, these numbers represent lower-bound estimates on how strongly gene conversion can slow down sequence divergence.
Median dS values for intra- and interspecific paralogs
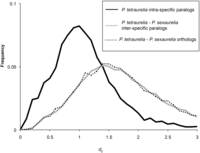
Frequency distributions of synonymous site divergence (dS) for intra- vs. interspecific paralogs and orthologs. Only comparisons of P. tetraurelia with P. sexaurelia are shown. All other species comparisons are shown in Table 3 and Supplemental Figure 2.
Because gene conversion can extend past the coding sequence of genes, we also used a method that detects abnormally high similarity in the noncoding DNA flanking each pair of paralogs (Evangelisti and Conant 2010). We extracted noncoding sequences upstream of and downstream from paralogs, aligned them, and computed an alignment score (Methods). To discriminate conservation caused by selection on noncoding sequence (typically regulatory elements) from sequence identity resulting from gene conversion, we compared the alignment scores of intraspecific paralogs to those of interspecific paralogs, assuming that gene conversion should result in an elevated score for intraspecific paralogs. The distribution of alignment scores was significantly biased toward higher values for intraspecific paralogs when compared to that of interspecific paralogs (P < 0.01), suggesting that gene conversion extends to the noncoding regions flanking paralogous genes. We also observed that genes with flanking regions having an intraspecific alignment score higher than their interspecific alignment score had significantly reduced dS and elevated GC (both P < 0.01). Because reduced dS and elevated GC content are typical of gene conversion, we interpret these observations as additional evidence supporting our conclusion that flanking noncoding regions successfully detected cases of gene conversion. Finally, we investigated conversion-tract lengths by identifying the longest stretches of identity between paralogs. The median of these tract lengths is 125 bp in P. biaurelia, 108 bp in P. tetraurelia, and 106 bp in P. sexaurelia (Supplemental Fig. 3), a pattern that mirrors the dS estimates above, suggesting a history of more frequent gene conversion between P. biaurelia paralogs than in the other two species.
We note that frequent inter-paralogs gene conversion could explain the observed positive correlation between GC content and gene retention. Indeed, regions of high meiotic recombination rate tend to be GC-rich because of GC-biased gene conversion (Galtier et al. 2001; Galtier 2003; Duret and Galtier 2009), a process that is likely to be operating in Paramecium (Duret et al. 2008). Therefore, paralogs in GC-rich regions are more likely to undergo meiotic recombination, which has the potential to oppose gene loss, as explained above. To test whether the correlation between GC content and gene retention reflects selective pressures on coding sequences (as proposed in the previous paragraph) or is the consequence of a higher meiotic recombination rate in GC-rich regions, we measured the correlation between gene retention and intronic GC content (after removing the first and last three nucleotides of each intron, as they are under selective constraint to maintain splice sites [Jaillon et al. 2008]) as well as for the first, second, and third positions in codons separately (GC1, GC2, and GC3, respectively). Under the selection model, the correlation is expected to disappear when looking specifically at sites under weak selective pressure, such as introns and the third position of codons. Although the signal is weaker, we still observe a positive correlation between gene retention and both GC3 and intronic GC in all three aurelia species (Supplemental Fig. 4), supporting the hypothesis that gene conversion promotes the retention of post-WGD paralogs in Paramecium. We also investigated the possibility that the positive correlation with GC3 is caused by biased codon usage. We derived a list of codons whose relative synonymous codon usage (RSCU) (Sharp and Li 1987) is higher in highly expressed genes. We found that 68%, 46%, and 57% of these codons use a G or C nucleotide at their third position in P. tetraurelia, P. biaurelia, and P. sexaurelia, respectively. Therefore, it is possible that biased codon usage partially explains the observed correlation between gene retention and GC3 in P. tetraurelia and P. sexaurelia. However, the fact that we still observe the same correlation in P. biaurelia, and because there is no reason to think that intronic GC content reflects selective pressures, the observed correlations suggest an impact of gene conversion on post-WGD gene retention.
At the first and second positions of codons, both purifying selection and GC-biased gene conversion act in the same direction, which might explain why the pattern is stronger. It is also important to note that, while selection on the coding sequence is still an ongoing process, meiotic recombination between paralogs was mostly confined to the early stages following the WGD. Therefore, it is not surprising that the signal for the impact of meiotic recombination on gene retention is weaker than that of selection on the coding sequence.
Finally, we considered the possibility that frequent meiotic recombination between paralogs increases the GC content of retained genes via GC-biased gene conversion, therefore creating the observed correlation between GC and retention. Under this model, high GC content would be a consequence of gene retention. However, we observed that the correlation between GC content and gene retention holds when using the ancestral (pre-WGD) GC content (McGrath et al. 2014), indicating that the higher GC content of retained genes is the cause and not the consequence of their retention, making this model unlikely.
Timing of WGD and speciation events
Because gene conversion between WGD-derived paralogs is widespread in Paramecium, using sequence divergence between paralogs from only one species downwardly biases the estimated age of the duplication. To eliminate this problem, we used sequence divergence between interspecific paralogs, because such paralogs do not have the opportunity to undergo conversion after lineage separation. The median dS between P. tetraurelia–P. sexaurelia interspecific paralogs is 1.7, which is significantly higher than the median dS of intraspecific paralogs in P. tetraurelia (median = 1.0; P < 0.001) (Table 3; Supplemental Fig. 2), indicating that the most recent WGD is ∼1.7× older than previously thought. Interestingly, the dS distribution of P. tetraurelia–P. sexaurelia interspecific paralogs is not statistically different from that of P. tetraurelia–P. sexaurelia orthologs (Fig. 6, plain gray line vs. dotted line, P = 0.2, Wilcoxon rank sum test), consistent with P. sexaurelia diverging from the lineage leading to P. biaurelia–P. tetraurelia immediately following, or concurrent with, the most recent WGD. Thus, the data are consistent with the hypothesis that the most recent WGD itself initiated the aurelia species radiation (Aury et al. 2006).
Using an estimate of the mutation rate in P. tetraurelia of 2.64 × 10−11 mutations per site per cell division (Sung et al. 2012), we estimate that the most recent WGD (and the P. sexaurelia–P. bi/tetraurelia speciation) occurred ∼32 × 109 cell divisions ago. This translates into ∼320 million years, assuming a conservative estimate of 100 cell divisions per year. Because both the mutation rate and the average number of cell divisions per year could have significantly changed since the WGD (although the division time remains roughly the same for all species of the complex), our margin of uncertainty on this estimation is wide. However, it seems clear that the most recent WGD occurred many tens of millions of years ago, which is remarkable when one considers that the different aurelia species are morphologically indistinguishable (Sonneborn 1975).
The pattern of gene losses favors a model of autopolyploidy over allopolyploidy
WGDs resulting from allopolyploidization events (hybridization of two species) are characterized by immediate differences in the paralogous chromosomes, which might lead to bias in the gene retention pattern, i.e., paralogs from one of the two species being more likely to be retained than those from the other species. Such asymmetrical loss patterns have been observed in Arabidopsis (Thomas et al. 2006), maize (Woodhouse et al. 2010), and rice (Wu et al. 2008). On the other hand, in the case of autopolyploidy, paralogous chromosomes are initially identical and are expected to be unbiased in their pattern of gene loss. We investigated whether duplicate loss was randomly distributed between the two paralogous scaffolds in each aurelia species by focusing on scaffolds with at least 50 pre-WGD predicted genes. We found significant asymmetrical gene loss in 11%, 4%, and 4% of these scaffolds, respectively, for P. tetraurelia, P. biaurelia, and P. sexaurelia (Supplemental File 12). The fact that only a minority of scaffolds show asymmetrical gene loss supports the hypothesis of an autopolyploidization event being responsible for the most recent WGD in Paramecium. Interestingly, while there is high similarity between which scaffold pairs have experienced asymmetrical loss between P. biaurelia and P. tetraurelia, there is little overlap in which scaffolds are asymmetrical between P. tetraurelia and P. sexaurelia. This suggests that the gene-loss asymmetry was introduced relatively recently, after the divergence of these two species, making it unlikely that the asymmetry reflects an allopolyploidy event. The small excess of asymmetrical gene loss observed here could have been caused by physical clustering of functional linked genes along chromosomes, as observed in human, yeast, and Arabidopsis (Makino and McLysaght 2012).
Discussion
In this study, we have examined duplicate-gene retention, loss, and evolution in three Paramecium lineages following a whole-genome duplication. We found that different species sharing the same WGD can show significant variation in the fraction of retained duplicated genes, resulting in large differences in the number of genes between these species. Despite these differences, some global trends of WGDs emerge, with most gene functional categories showing similar patterns of loss across species and parameters such as gene-expression level being universally associated with post-WGD gene retention. This study also provides evidence for widespread gene conversion between paralogs derived from a WGD. Although most paralogs are now divergent enough that very few of them still undergo frequent gene conversion in Paramecium, this process was probably very active shortly after the WGD. Interestingly, our analysis suggests that gene conversion has actively promoted paralogs’ retention in Paramecium.
We have refined the estimated age of the WGD and speciation events, concluding that these events were much older than previously thought, and showing that—although the fate of most duplicated genes is eventual loss—a large number of paralogs can be preserved in two copies for millions of years after a WGD. Our conclusion that the first speciation events in the aurelia complex occurred immediately after—or concomitant with—the most recent Paramecium WGD, coupled with our observation that divergent gene loss was frequent in the early times following the WGD, makes divergent gene losses a strong candidate for the emergence of the species that formed the aurelia complex. While this passive mechanism of speciation only requires differential gene losses, it has been argued that WGDs can also lead to phenotypic innovations and increased morphological complexity (Ohno 1970; Freeling 2008). Indeed, it has long been suggested that duplicated genes provide the raw material for evolving new functions, so that the thousands of duplicated genes created by a WGD represent unique opportunities to increase organisms’ complexity (Freeling 2008). For example, the two rounds of WGDs that occurred at the basis of vertebrates, as well as the extra round of WGD specific to the teleost fishes lineage, have been linked to the evolutionary success and morphological complexity of these lineages (Holland et al. 1994; Meyer and Van de Peer 2005; Freeling and Thomas 2006). WGDs are also suspected to have played an important role in the increased complexity of land plants (Van de Peer et al. 2009) and in the development of a lifestyle based on glucose fermentation in yeast (Thomson et al. 2005; Woolfit and Wolfe 2005). In contrast to these examples, the Paramecium WGDs do not seem to have fueled phenotypic innovations. Despite the fact that some of these species diverged several million years ago and have considerable gene number differences (up to 4000 genes), they are still morphologically indistinguishable (Sonneborn 1975). Although it is possible that some phenotypic innovations have been overlooked and remain to be discovered, it is striking to observe that, despite creating thousands of new genes, these two rounds of WGDs did not result in any obvious phenotypic innovation in the P. aurelia lineage.
Methods
Genomic DNA preparation, extraction, sequencing, and assembly
Roughly 2 L of P. biaurelia (strain V1-4) and P. sexaurelia (strain AZ8-4) were grown in Wheat Grass Powder (Pines International) medium (Aury et al. 2006), starved, and Paramecium cells purified away from bacteria by filtration over a 10-μm Nitex membrane. Macronuclei were isolated away from other cellular debris by gentle lysis of the cell membrane and sucrose density separation (Aury et al. 2006). DNA was extracted and purified using a CTAB protocol (Doyle and Doyle 1987). We obtained 8-kb-insert 454 GS FLX (Roche) mate-pair reads (12× coverage) and Illumina (Illumina) paired-end reads (30× coverage) for each species. Illumina and 454 reads for each genome were co-assembled using the Celera assembler (Miller et al. 2008).
RNA preparation, extraction, sequencing, and mapping
Roughly 1 L of P. biaurelia (strain V1-4) and P. sexaurelia (strain AZ8-4) were grown in Wheat Grass Powder medium to mid-log phase, and Paramecium cells were purified away from bacteria by filtration over a 10-μm Nitex membrane. Whole-cell RNA was isolated using TRIzol (Ambion) and the manufacturer’s suggested protocol for tissue culture cells. We obtained Illumina paired-end reads (∼1000 reads/gene) for each species. RNA-seq reads were mapped to each genome using Bowtie/TopHat (Langmead et al. 2009; Kim et al. 2013), retaining only reads that mapped unambiguously, and transcripts were predicted using Cufflinks (Trapnell et al. 2012). The abundance of each mRNA was predicted from a logarithm transformation of the FPKM (fragments per kilobase of exon per million fragments mapped) values reported by Cufflinks. Prior to the log transformation, all genes with a FPKM of 0 were given the value 0.1.
Paramecium gene annotations
P. biaurelia and P. sexaurelia genes were identified using a combination of RNA-seq data, BLAST hits to P. tetraurelia predicted genes (v1.68), and two de novo gene prediction programs, Augustus and EuGene. P. tetraurelia predicted genes (v1.68) were downloaded from ParameciumDB (Arnaiz and Sperling 2011), and BLASTN (Camacho et al. 2009) was used to query each genome (E-value cutoff = 1 × 10−10) for related genes. Identified genomic regions were aligned with the corresponding P. tetraurelia gene using CAP (Huang and Madan 1999), and alignments were further refined using MUSCLE (Supplemental Table 5; Edgar 2004).
A subset of 6519 high-confidence (“Model fully confirmed with ESTs”) P. tetraurelia genes were downloaded from ParameciumDB, and putative genes from the BLAST procedure above that had been identified using these high-confidence genes as the query were considered high-confidence putative P. biaurelia (5880) and P. sexaurelia (4937) genes. After filtering for low-quality alignments and in-frame stop codons, and after removing one member of each pair of recent paralogs (to avoid overfitting the model), we trained Augustus (Stanke et al. 2008) for each species with a training set of 2331 P. biaurelia and 1111 P. sexaurelia genes. Augustus was then used to predict genes for each genome (Supplemental Table 5). Augustus-predicted genes were then merged with BLAST-predicted genes to create a set of gene models for use as hints when running EuGene (Supplemental Table 5). Using the original high-confidence putative P. biaurelia and P. sexaurelia genes as a test for accuracy, 72.9% of the high-confidence P. biaurelia genes and 70.6% of the high-confidence P. sexaurelia genes were predicted with correct start and stop codons in this merged Augustus/BLAST prediction, while an additional 27.0% of the P. biaurelia genes and 16.5% of the P. sexaurelia genes overlapped with genes from the Augustus/BLAST prediction.
A version of EuGene (Schiex et al. 2001; Foissac et al. 2008) trained on P. tetraurelia (O Arnaiz and L Sperling, in prep.) was run on the P. biaurelia and P. sexaurelia genomes using the Cufflinks transcripts and the Augustus/BLAST-predicted genes (from above) as evidence. This combination was shown to give the highest accuracy according to comparisons with the high-confidence P. biaurelia and P. sexaurelia data sets. In P. biaurelia, 75.7% of the high-confidence genes were accurately predicted with the correct start and stop codons, and an additional 23.7% overlapped with predicted genes. In P. sexaurelia, 72.8% of the high-confidence genes were accurately predicted with the correct start and stop codons, and an additional 27.1% overlapped with predicted genes (Supplemental Table 5). While the high-confidence genes are likely to be a somewhat biased subset of all genes, these figures suggest that <1% of genes are unrepresented in our final annotations. Functional predictions for genes were annotated using PANTHER (Supplemental Table 5; Mi et al. 2012). We used the published P. tetraurelia genome (Aury et al. 2006) and current annotations (v1.85) available at ParameciumDB (Arnaiz and Sperling 2011) for final comparisons with the P. biaurelia and P. sexaurelia genomes.
Alignment of paralogous segments within each species
The procedure for identification of paralogous genomic segments within each species was modified from Aury et al. (2006). BLASTP was used to identify homology between proteins within each genome (E-value cutoff = 1 × 10−10). Sliding windows of 20 genes were then analyzed, and if >40% of the genes in this window had reciprocal best BLAST hits (RBHs) to the same scaffold in the genome, a paralogous block was created. Contiguous windows where the same two paralogous scaffolds were involved in the block were merged to create the largest possible paralogous blocks. Additional matches were added when a “singleton” gene (a gene without a RBH within the paralogous block) was represented among the top 10 BLAST hits of another singleton gene in the same paralogous block. The full set of paralogous blocks and the included genes for each species can be found in Supplemental Files 3–5.
To ensure that inferred gene losses were not biased due to assembly gaps, we also created a version of paralogous blocks where regions surrounding assembly gaps were removed. Specifically, genomic regions around gaps up to the next duplicated gene were removed from the analysis. With this method, we assured that no genes were inferred as single-copy unless the paralogous segment between the next upstream and downstream duplicated genes was free of gaps (Supplemental Files 7–9).
Alignment of orthologous segments between species
Identification of orthologous genomic segments between P. tetraurelia and P. biaurelia was identical to the method for identification of paralogous segments above, with BLASTP identifying homologous proteins between the two species. Because of the similar divergences between P. tetraurelia–P. sexaurelia orthologs and P. tetraurelia–P.sexaurelia paralogs, however, the use of RBHs to identify orthologous blocks between these two species meant that each P. tetraurelia genomic segment matched two P. sexaurelia genomic segments (the true orthologous segment and the interspecific paralogous segment), and vice versa. To differentiate between orthologous and paralogous blocks between these two species, the number of genes in common between each interspecific pair of genomic segments was tallied, and orthology was assigned to maximize the number of genes that orthologous blocks shared (minimizing the number of inferred gene losses between orthologous blocks). The full set of orthologous blocks between P. biaurelia–P. tetraurelia and between P. tetraurelia–P. sexaurelia can be found in Supplemental Files 13 and 14.
Once orthologous blocks were identified between P. tetraurelia and each newly sequenced genome (P. biaurelia and P. sexaurelia), these data sets were merged, along with the paralogy information for each genome, to give the full set of orthologous/paralogous blocks across all three species, each block comprising six genomic segments (two paralogous segments from each species). During the merging, additional BLASTP matches between genes in P. biaurelia and P. sexaurelia that are not present in P. tetraurelia were identified and annotated. The full set of orthologous/paralogous blocks across all three species can be found in Supplemental File 10.
Phylogeny of P. biaurelia, P. tetraurelia, and P. sexaurelia and estimates of dN/dS
Estimates of dN and dS for pairs of orthologs and paralogs were obtained by aligning amino acid sequences using MUSCLE (Edgar 2004) and then reverse translating back into nucleotide sequences. dN and dS were then calculated from the nucleotide alignments using codeml in PAML (Yang 2007).
For Figure 1, a maximum likelihood phylogeny was constructed of the three aurelia species with P. caudatum as an outgroup. One hundred MUSCLE alignments of ortho-paralog families were randomly selected, and one set of orthologs (including P. caudatum, where available) from each ortho-paralog family was included. These amino acid sequences were concatenated, adding gaps where orthologs from individual species were missing due to gene loss. A phylogeny was created from the resulting alignment using PhyML (Guindon et al. 2010). PhyML was also used to generate 100 bootstrap replicates, and the consensus tree with bootstrap values is given in Figure 1.
Identifying tandem duplications
We conducted an all-vs.-all BLASTP search within each genome, keeping the top five hits with an E-value < 1 × 10−10. If a gene had a BLAST hit against a neighboring gene, we considered it a tandem duplicate. When available, we used the ortholog/paralog information to determine whether a tandem duplicate dated from before the recent WGD or before a speciation event, or whether it was unique to a species (no aligned orthologs or WGD paralogs).
Examining patterns of gene conversion using alignments of noncoding sequences
We extracted up to 250 bp of noncoding DNA immediately upstream of (5′) and downstream from (3′) each gene. For cases where the intergenic region between a gene and its neighbor was between 100 and 500 bp, we split the intergenic region in half, assigning half to each gene. Cases where the intergenic region was shorter than 100 bp were removed from the analysis. 5′ and 3′ flanking sequences for pairs of paralogs were aligned separately using MUSCLE (Edgar 2004) and were scored using Kimura’s two-parameter model (Kimura 1980).
Investigating asymmetrical gene loss between paralogous scaffolds
For every pair of intraspecific paralogous scaffolds with at least 50 ancestral, pre-duplicated genes, we counted the number of genes lost from each scaffold and then calculated the proportion of ancestral genes lost on each scaffold (# of genes lost/# of ancestral genes). We used a χ2 statistic to test the significance of the difference in retention rates between the two scaffolds. We applied a Bonferroni correction for multiple testing to the P-values.
Assignment of gene losses along the evolutionary tree
The list of aligned orthologous/paralogous blocks across all three species was used to classify each gene family into one of the 63 possible gene loss/retention outcomes (Supplemental Fig. 1). We used parsimony to assign gene losses along a branch for each outcome and then used the number of gene families falling into each outcome to calculate the number of losses on each branch. We repeated the analysis removing all solo genes (Supplemental Fig. 4). We used the median dS values between orthologs and paralogs and split them equally between the two branches to determine dS values for each branch. We then determined the percent gene retention for each species and ancestor in the tree by taking the number of original duplicates in the analysis, subtracting the number of losses along the tree up to that point, and dividing by the number of original duplicates. Note that because species can lose both duplicates, this is a gene retention rate, not strictly a duplicate retention rate.
Functional analysis of retained duplicates vs. single-copy genes
GO terms assigned by PANTHER and the gene retention data from above were used to analyze functional categories of genes with an overretention or underretention of duplicates. The percentage of ancestral genes still duplicated for each GO term was compared to the percentage of ancestral genes still duplicated for all other GO terms within the same GO category (Molecular Function, Biological Process, or Cellular Component) with a χ2 test. Significance was determined after correcting for multiple testing by controlling the FDR at 5% (Benjamini and Hochberg 1995).
Relationship between GC content, expression level, and duplicate gene retention
Expression level and GC values were divided into bins. Genes were sorted into bins based on their variable values and then the percent retention for each bin was calculated by the following equation: ½ duplicate genes ÷ (½ duplicate genes + singleton genes). A weighted least-squares regression was then performed for retention on GC or expression level. To determine whether both expression level and GC content were independent predictors of retention, we completed a logistic regression for each species with duplication status (1 for duplicated, 0 for singleton) as the binary dependent variable and GC content and expression level of each gene as the predictor variables.
Data access
The Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank (http://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA252371 (P. biaurelia) and PRJNA252373 (P. sexaurelia). Sequences and annotations can also be downloaded from ParameciumDB (http://paramecium.cgm.cnrs-gif.fr/download/species/).
Acknowledgments
The authors thank O. Arnaiz and L. Sperling for sharing the EuGene annotation pipeline trained on P. tetraurelia. Funding for this work has been provided by an NSF Graduate Research Fellowship to C.L.M., NIH GCMS Training Grant T32 GM007757 to C.L.M., and NSF grant EF-0328516-A006 to M.L. This research includes work supported by the National Science Foundation under Grant no. ABI-1062432 to the National Center for Genome Analysis Support at Indiana University.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.173740.114.
- Received February 7, 2014.
- Accepted July 8, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








