
Random replication of the inactive X chromosome
- 1Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA;
- 2Program in Medical and Population Genetics, and Stanley Center for Psychiatric Research, Broad Institute of Harvard and MIT, Cambridge, Massachusetts 02142, USA
Abstract
In eukaryotic cells, genomic DNA replicates in a defined temporal order. The inactive X chromosome (Xi), the most extensive instance of facultative heterochromatin in mammals, replicates later than the active X chromosome (Xa), but the replication dynamics of inactive chromatin are not known. By profiling human DNA replication in an allele-specific, chromosomally phased manner, we determined for the first time the replication timing along the active and inactive chromosomes (Xa and Xi) separately. Replication of the Xi was different from that of the Xa, varied among individuals, and resembled a random, unstructured process. The Xi replicated rapidly and at a time largely separable from that of the euchromatic genome. Late-replicating, transcriptionally inactive regions on the autosomes also replicated in an unstructured manner, similar to the Xi. We conclude that DNA replication follows two strategies: slow, ordered replication associated with transcriptional activity, and rapid, random replication of silent chromatin. The two strategies coexist in the same cell, yet are segregated in space and time.
DNA synthesis in eukaryotic cells initiates from many origins of replication. Only a small subset of the available origins is used within a given cell cycle, and the specific origins that are utilized differ from cell to cell. Nevertheless, at large scales, genomic loci replicate in a reproducible temporal order that is thought to result from variations along the genome in the rates and preferred activation timing of replication origins. A notable exception to programmed genome replication occurs in early embryonic development in Xenopus laevis and Drosophila melanogaster. During cleavage cell divisions, when there is no transcriptional activity, DNA replication initiates from closely spaced loci that are located randomly with respect to the DNA sequence, and is completed in a very short time (several minutes, compared with hours in adult cells) (Hyrien and Mechali 1993; Sasaki et al. 1999). DNA replication becomes organized, with replication origins activated at specific times and locations, only at the mid-blastula transition, concomitant with the onset of zygotic transcription (Hyrien et al. 1995; Sasaki et al. 1999). The observation that structured replication is established just as transcription commences suggests that a strict replication program may be required for the regulation of gene activity. However, random replication has not been observed in mammals or outside the context of embryonic development.
In mammals, developmentally regulated, chromosome-wide transcriptional silencing occurs in the process of X chromosome inactivation (XCI). XCI is a dosage compensation mechanism in which one of the two X chromosome copies in females is transcriptionally inactivated (Lyon 1961). The inactive chromosome is randomly chosen and subsequently clonally maintained through an epigenetic mechanism that involves coating of the chromosome by the noncoding RNA XIST (X-inactivation specific transcript), DNA hypermethylation, histone modifications, and other chromatin marks (for review, see Lee 2011). The Xi localizes to the perinucleolar compartment during mid-to-late S phase (Zhang et al. 2007) and adopts a three-dimensional conformation that was recently shown to be random in mice (Splinter et al. 2011). The Xi replicates later in S phase than the Xa (Gilbert et al. 1962; Morishima et al. 1962; Hansen et al. 1996, 2010; Chadwick and Willard 2003; Heard and Disteche 2006). Whether the Xi follows a defined replication timing program remains unknown, although early microscopy studies suggested that its replication differs from that of the Xa (Gilbert et al. 1962; Morishima et al. 1962; Willard and Latt 1976; Schempp and Meer 1983).
Results
We recently described the profiling of genomic DNA replication in lymphoblastoid cell lines (LCLs) from two father–mother–daughter trios by sequencing DNA from FACS-sorted G1 and S phase cells. Replication timing was inferred from the fluctuations in the abundance (read depth) of DNA sequences along chromosomes: The earlier a locus replicates, the higher its abundance in genomic DNA in S phase cells (Koren et al. 2012). We next sought to determine the replication profiles of each of the 46 chromosomes individually. We identified all heterozygous SNPs (median spacing of ∼800 bp) from the genome sequence of these cell lines (The 1000 Genomes Project Consortium 2010) and assigned sequence reads to individual chromosomal copies by using inheritance in the trios to determine chromosomal phase for each SNP allele (Methods). We thus obtained replication profiles for each of the 46 chromosomes, and were further able to distinguish the active from the inactive X chromosome (Methods; Supplemental Fig. S1).
The timing of Xi replication was mostly distinct from that of all other chromosomes: 75% of the Xi commenced replication after almost 75% of the rest of the genome had already completed replication (Fig. 1A). Intriguingly, despite being the last chromosome to initiate replication and having the latest average replication time, the Xi still completed replication no later than the rest of the genome (Fig. 1A), suggesting that the Xi replicates more rapidly than other chromosomes do. A direct analysis revealed that the bulk of Xi replication (defined by the interquartile range of replication timing) completed approximately twice as fast as any other chromosome, and almost three times faster than the bulk of Xa replication (Fig. 1B).
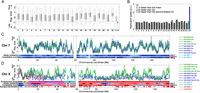
Replication dynamics of the 46 human chromosomes. (A) Distribution of replication timing for each of the 46 chromosomes in lymphoblastoid cell line (LCL) NA19240. For each chromosome, thin vertical lines show the distribution of replication times; thick bars show the 25th and 75th percentiles; and the horizontal line shows the median. (Gray) Maternally inherited chromosomes; (black) paternally inherited chromosomes; (green) the active X chromosome (Xa); (blue) the inactive X chromosome (Xi). Dashed horizontal line is the Xi 25th percentile. The Xi replicated at a time separable from most of the genome. Results for LCLs from the other two females were similar (data not shown). (B) Replication speed (the inverse of the replication time span) of the bulk (interquartile range) of each chromosome in cell line NA19240. The dashed horizontal line represents the autosomal average. Results for LCLs from the other two females were similar (data not shown). Note that the high-GC content chromosome 22 is not shown since its values were unreliable due to relatively poorer data quality. (C) Smoothed chromosome 7 replication profiles of LCLs derived from three females (and one experimental replicate; green and blue) and one male (cyan). The replication profile was similar among individuals and between homologous chromosomes. (Mat) maternal; (Pat) paternal; (1,2) homologous chromosome copies for which parent-of-origin is unknown (Supplemental Fig. S1); [(2)] experimental replicate. Replication time is normalized as a Z-score (Koren et al. 2012). Allelic similarity is the similarity in replication timing between the homologous chromosome copies; correlation score represents the similarity of replication pattern between the homologs (Methods). (Gray vertical lines) Centromere. Results for other autosomes were similar (data not shown). (D) Smoothed replication profiles of chromosome X, showing the delayed, unstructured, and variable replication timing of the Xi relative to the consistent and structured Xa. The X (cyan) and Y (red) chromosomes of two males are shown; the correlation between the two Y chromosomes was r = 0.89. Also shown are the tendency of genes to escape X inactivation, in fractional units, and the X chromosome evolutionary strata. (P) Pseudoautosomal region; (S) stratum. PAR1 and strata 4 and 5 showed the typical autosomal signature of allelic similarity. No other regions on the Xi appeared to have any significant replication structure at the sensitivity level of detection of our method (∼0.5 standard units of replication timing, corresponding to ∼10% of the replication time span—see panel C). XIST replication timing is considered more specifically in Supplemental Figure S3. See Supplemental Figure S2 for more detailed images of X chromosome replication, including replication profiles obtained with higher coverage data for specific regions of the chromosome.
The replication profiles of the autosomes were visually indistinguishable between homologous chromosome copies, between different individuals, and between experimental replicates (Fig. 1C; Supplemental Fig. S2). We verified this statistically using a correlation analysis, which revealed highly correlated replication timing in each of these comparisons (mean r = 0.85) (Fig. 2A). Analysis of the X chromosome, however, revealed a strikingly different pattern. Xa replication profiles were similar among females and consistent between experimental replicates (Figs. 1D, 2A); they were also similar to the replication profiles of the X chromosome in males (mean r = 0.85). In contrast, replication of the inactive X chromosomes was far less similar (mean r = 0.39) to that of the active copies, with only a few zones of limited similarity (Figs. 1D, 2A). The Xi replication patterns also differed among the three females (mean r = 0.37) and between experimental repeats (r = 0.39, compared with r = 0.79 for the Xa). Furthermore, the “diploid” X chromosome profiles (which represent a composite of the two chromosomal copies not limited by SNPs) closely resembled the Xa but not the Xi profiles. Taken together, these results suggest that the Xi does not replicate according to a spatially structured program.
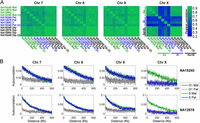
Structure and correlations of chromosomal replication profiles. (A) Correlation of replication timing for pairs of homologous chromosomes, within and between individuals and experimental replicates, shown as correlation matrices. For the autosomes, replication profiles were similar among individuals and between chromosome homologs in the same individual. Replication of the Xa was also consistent among individuals; however, replication of the Xi was different from the Xa, was uncorrelated among individuals, and was inconsistent between experimental replicates. (Mat) Maternal; (Pat) paternal; (1,2) homologous chromosome copies for which parent-of-origin is unknown (Supplemental Fig. S1); (Dip) diploid (aggregated across the two homologous chromosome copies); [(2)] experimental replicate. (B) Autocorrelations of DNA replication timing. The highly structured replication of the autosomes and the Xa is visible as long-range autocorrelation of DNA copy number in S phase genomes (but not in control, G1 phase genomes). In contrast, the inactive X chromosome shows no more autocorrelation in S phase than in G1 phase cells. Results for NA12892 were similar (data not shown). For both A and B, the 8-Mb left-distal part of the X chromosome was excluded from the analyses (see text), and results for other autosomes were similar (data not shown).
To more formally test for a replication structure on the Xi, we used autocorrelation analysis to evaluate the correlation between the replication timing of distinct sites as a function of the physical distance between them (Methods). We separately analyzed autocorrelation of read depth in S phase cells, which represents the continuous process of DNA replication, and in G1 cells, a control that will manifest any technical fluctuations in sequence read depth (Koren et al. 2012). As expected, G1 phase cells exhibited only a slight positive autocorrelation (Fig. 2B), explained by the known effect of GC content on read depth. On the other hand, S phase DNA abundance across the autosomes and the Xa exhibited a strong autocorrelation for distances of several hundred kilobases, indicating a spatially structured program (Fig. 2B). Strikingly, in all three females, S phase DNA abundance of the Xi had autocorrelation that was no stronger than that of G1 DNA, indicating that the Xi replication pattern resembles a random process (Fig. 2B).
We then evaluated the extent of replication randomness along the X chromosome and among different evolutionary domains of the X chromosome. Mammalian sex chromosomes evolved from a pair of autosomes through a series of inversions on the Y chromosome that led to loss of recombination and subsequent sequence divergence between the X and Y chromosomes. Five discrete evolutionary strata of progressive X–Y evolutionary divergence have been observed. Sequence homology and recombination activity are retained at the 2.7-Mb-long pseudoautosomal region 1 (PAR1) and the 0.3-Mb-long PAR2 on the left and right ends, respectively, of the X and Y chromosomes (Fig. 1D; Ross et al. 2005).
We analyzed the average difference in replication timing and the consistency of replication pattern between the two copies of each chromosome along their entire lengths. Autosomes exhibited a characteristic signature of consistent replication structure and timing for each pair of chromosome homologs (Fig. 1C). In contrast, the X chromosome exhibited low allelic similarity and correlation throughout its length, consistent with a cis-effect on replication timing and structure (Fig. 1D). The notable exception was the ∼8-Mb region on the distal short arm of the X chromosome, which contains PAR1 and evolutionary strata 4 and 5. This region replicated relatively early and exhibited replication patterns typical of autosomes, including similar replication timing and structure for the homologous chromosome pairs (Xa and Xi) (Fig. 1D; Supplemental Figs. S2, S3). Remarkably, the replication pattern of the corresponding 8-Mb area on the left end of the Y chromosome also showed a similar replication pattern, even though X–Y syntenic homology extends only to the PAR1 region (Fig. 1D; Supplemental Fig. S2). We conclude that an ∼6-Mb region beyond PAR1 replicates in an ordered and coordinated way on the Xa, Xi, and Y chromosomes. Intriguingly, the same region also contains a cluster of genes that escape X inactivation and are expressed from both the Xa and Xi (Fig. 1D; Carrel and Willard 2005). This area is thought to have been pseudoautosomal in recent evolutionary history (Ross et al. 2005), and we propose that it is still effectively pseudoautosomal from an epigenetic point of view, i.e., that it retains similar epigenetic determinants of replication timing on the X and Y chromosomes and is not subject to dosage compensation on the X chromosome.
The observed lack of replication structure on Xi could arise from features specific to X inactivation, or could represent a general property of late-replicating chromatin on all chromosomes. To distinguish between these possibilities, we partitioned autosomal genomic segments into eight groups based on their replication timing and analyzed the autocorrelation along genomic segments in each group (Fig. 3). Autocorrelation of replication timing decreased as S phase progressed, indicating a gradual loss of replication structure. Furthermore, autocorrelation decreased along with gene density, transcription levels, and the proportion of open chromatin in the genomic segments being replicated (Fig. 3). Thus, random replication is a general property of late-replicating, transcriptionally inactive chromatin, and cells transition from slow, organized replication to fast, random replication as S phase progresses. The increase in replication randomness as S phase progressed was sufficient to account for most, if not all, of the randomness of the Xi (Supplemental Fig. S4).
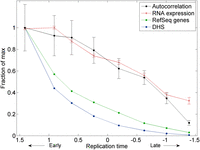
Random, unstructured replication of late-replicating, transcriptionally inactive genomic segments on the autosomes. Autosomal segments were divided into eight groups based on their replication timing. Shown for each group are the autocorrelation of replication timing at a ∼2-kb scale (averaged over all six individuals profiled; Methods), the number of RefSeq genes, average level of gene expression (measured by number of RNA-seq reads), and density of DNase I hypersensitive sites (DHS) representing the 5% most open chromatin regions in lymphoblastoid cell lines. Error bars, SE. All data are scaled relative to the maximal values for each data type.
Discussion
In this study, we have determined for the first time the detailed replication dynamics of the human active and inactive X chromosomes. Consistent with single-cell microscopy studies (Gilbert et al. 1962; Morishima et al. 1962), the Xi replicated much later than the Xa. Furthermore, we found that the Xi did not follow any defined temporal replication pattern: The replication profiles of Xi chromosomes were inconsistent among females and between experimental repeats, and did not show a continuous spatial pattern as measured by an autocorrelation analysis. A single exception was an 8-Mb region on the distal short arm of the X chromosome. Although the spatial and temporal resolution of our method might not be sufficient for the identification of small patches of ordered replication along the inactive X chromosome, these are expected to be limited to—at most—submegabase scales, and to a fraction of the time span within the already confined late replication of the Xi. The random pattern of replication of the Xi that we observe is to be distinguished from the previously described “stochastic” pattern of origin activation: While the latter still follows probabilistic patterns that give rise to structured replication profiles in population measurements (and is thought to follow time constraints in mammalian cells), Xi replication appears to be literally random, giving rise to population-level timing patterns that are not more structured than noise is.
These results provide the first demonstration of random replication in somatic human cells. Replication of the Xi was rapid, random, and associated with transcriptional quiescence, recapitulating all of the properties of early embryonic replication in frogs and flies. Random, rapid replication could be explained by stochastic firing of a large number of closely spaced origins (Fig. 4), as observed in frog and fly embryos (Hyrien and Mechali 1993; Hyrien et al. 1995; Sasaki et al. 1999). The gradual loss of replication structure as S phase progresses (Fig. 3) is consistent with elevated origin initiation rates at later times during S phase, as observed in single-molecule analyses in frogs (Herrick et al. 2000), human cells (Guilbaud et al. 2011), and fission yeast (Patel et al. 2006), and as predicted by mathematical models (Rhind 2006). Such an increase in firing probabilities as S phase progresses—which at the extremity of very late S phase results in random replication—has been suggested to be a principle property of DNA replication in eukaryotes (Goldar et al. 2009).
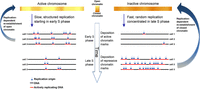
A model of replication structure regulation. Replication origins in regions that are transcriptionally active and have an open chromatin structure are activated at specified times (albeit with intercell variability in origin locations), giving rise to discrete domains that replicate in a synchronous manner. As S phase progresses, origin specification is gradually lost such that regions that replicate late use multiple origins are activated in a random manner, giving rise to an unstructured yet rapid replication pattern. In turn, dynamic changes in the activity of chromatin modifying enzymes over the course of S phase enable the preferential establishment of open or closed chromatin structures at particular genomic regions. The need to preserve chromatin structure during DNA replication may explain why cells sacrifice speed to achieve a specific temporal order of replication of transcriptionally active DNA.
Our results suggest that DNA replication in eukaryotes can proceed very rapidly without a particular order. In contrast, in the presence of transcriptional activity cells utilize a structured replication program that comes at a substantial cost in replication speed. This tradeoff suggests a need to coordinate replication timing with transcription, or a role for replication timing in preserving the epigenetic information that is required to regulate transcription. Such a role could be accomplished via dynamic changes in replication-associated chromatin modifying activities as chromatin is reassembled on newly synthesized DNA (Fig. 4; Hiratani et al. 2009). It has previously been suggested that the replication timing of the Xi is important for the inheritance of its epigenetic state (Chadwick and Willard 2003; Heard and Disteche 2006). DNA replication timing could potentially contribute to the transmission of epigenetic information across cell divisions, supporting the epigenetic maintenance of X chromosome inactivation and of chromatin states elsewhere in the genome.
Methods
Cell lines
Cell lines used were from two father–mother–daughter trios, one with European ancestry (CEU) and one with West African ancestry (YRI) (Supplemental Fig. S1). These cell lines show severe XCI skewing, in which the same copy of the X chromosome is inactive in >90% of the cells in a culture (McDaniell et al. 2010; Kucera et al. 2011). For both daughter cell lines, the paternally derived X chromosome was the clonally inactive copy (McDaniell et al. 2010; Kucera et al. 2011). We relied on the late replication of one X chromosome copy in the CEU mother (NA12892) as a marker for the Xi (Willard 1977) (an assignment also supported by the similarity to the Xi chromosomes in all other analyses we performed).
Replication timing profiles
Raw replication data was from Koren et al. (2012). For each individual, we generated a reference sequence based on the hg18 version of the human genome with all SNPs in that individual (The 1000 Genomes Project Consortium 2010) masked, and then aligned the raw replication sequence reads to that reference.
For each trio, we first identified all of the heterozygous SNPs in the daughter that were homozygous in at least one of the parents. We were thus able to assign each SNP allele to either a paternal or maternal origin. For the parents, heterozygous SNPs that were homozygous in the daughter were assigned as either transmitted or nontransmitted. Information of crossover location in the CEU parents (Fan et al. 2011) was used to switch the chromosomal assignment of consecutive SNPs, resulting in fully phased chromosomes, yet with no parental assignment.
For each individual with fully phased chromosomes (the two daughters and the CEU parents), sequence reads that overlapped a phased SNP were used to generate a chromosomal copy specific replication timing profile as described in Koren et al. (2012). The number of X chromosome reads available in each cell line and each fraction are detailed in Supplemental Table S1. Non-chromosomal-copy-resolved (“diploid”) data are from Koren et al. (2012).
Correlations and autocorrelations
The smoothed data were used to calculate correlations. Calculations using the nonsmoothed S/G1 read coverage yielded similar results (data not shown).
To compare the overall similarity of replication profiles along chromosomes, we devised a “correlation score” metric (Fig. 1) as follows: We (1) averaged all of the correlations between the “inconsistent” patterns, i.e., between maternal copies and between maternal copies and the diploid data (all of the blue blocks in the chromosome X correlation matrix in Fig. 2A); (2) averaged all of the correlations between the “consistent” patterns, i.e., between paternal copies, between paternal and maternal copies, and between paternal copies and the diploid data (all of the green blocks in the chromosome X correlation matrix in Fig. 2A); (3) calculated the ratio of the correlations obtained in 1 and in 2. The correlation score was thereby calculated in 10-Mb windows centered every 1 Mb along each chromosome.
Allelic difference was calculated in 10-Mb windows centered every 1 Mb and averaged over the three females. Autocorrelations were calculated on the raw number of reads per SNP in the G1 and S phase data. For calculating autocorrelations along S phase, the ratio of the number of reads in the S phase to the number of reads in the G1 phase in each 100-bp window was extracted for the autosomes of each individual (including the repetition of NA19240). Windows within 1 Mb of a sequence gap and windows with values more than 10 times the median window value for each individual were removed from the data. The data was separated into eight fractions of increasing replication timing, with equal amounts of data in each fraction (hence, the fractions are not always separated by the same time difference; because autocorrelation values are sensitive to the amount of data analyzed, this was more reliable than separating the data to equal-time fractions), and autocorrelations were calculated. For plotting, the average autocorrelations in the first 5–50 windows for each individual were used. Consistent results were obtained when analyzing the allele-specific data or the data defined by windows with a constant number of reads in the G1 phase (Koren et al. 2012), when separating the genome to a different number of fractions, and when using larger window sizes (data not shown).
Features of the X chromosome
Data regarding escape from X inactivation were extracted from Carrel and Willard (2005) and converted to fractional units by dividing the number of Xi hybrids that showed escape by the total number of hybrids assayed. Gene coordinates were converted to hg18 coordinates. Only assayed genes were used. Locations of evolutionary strata were from Carrel and Willard (2005).
Additional data sets
Average and standard error of gene expression were based on RNA-seq data for CEU cell lines (Montgomery et al. 2010). DNase I hypersensitive sites (DHS) were from Degner et al. (2012) and represent the 5% most open-chromatin sites in the genomes of 77 lymphoblastoid cell lines.
Data access
Deep coverage data for selected X chromosome regions (Supplemental Fig. S2) have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP029958.
Acknowledgments
We thank Jeannie Lee and Chris Patil for helpful comments on the manuscript. This work was supported by startup funds from the Department of Genetics at Harvard Medical School.
Footnotes
-
↵3 Corresponding author
E-mail mccarroll{at}genetics.med.harvard.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.161828.113.
- Received June 7, 2013.
- Accepted September 23, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








