
Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions
- Nathan C. Sheffield1,2,
- Robert E. Thurman3,
- Lingyun Song2,
- Alexias Safi2,
- John A. Stamatoyannopoulos3,
- Boris Lenhard4,5,8,
- Gregory E. Crawford2,6,8 and
- Terrence S. Furey7,8
- 1Program in Computational Biology and Bioinformatics, Duke University, Durham, North Carolina 27710, USA;
- 2Institute for Genome Sciences & Policy, Duke University, Durham, North Carolina 27710, USA;
- 3Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA;
- 4Bergen Center for Computational Science and Sars Centre for Marine Molecular Biology, University of Bergen, N-5008 Bergen, Norway;
- 5Department of Molecular Sciences, Institute of Clinical Sciences, Faculty of Medicine, Imperial College London, London, United Kingdom; and MRC Clinical Sciences Centre, London W12 0NN, United Kingdom;
- 6Department of Pediatrics, Division of Medical Genetics, Duke University, Durham, North Carolina 27710, USA;
- 7Department of Genetics and Department of Biology, Carolina Center for Genome Sciences, Linberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, North Carolina 27599, USA
Abstract
Regulatory elements recruit transcription factors that modulate gene expression distinctly across cell types, but the relationships among these remains elusive. To address this, we analyzed matched DNase-seq and gene expression data for 112 human samples representing 72 cell types. We first defined more than 1800 clusters of DNase I hypersensitive sites (DHSs) with similar tissue specificity of DNase-seq signal patterns. We then used these to uncover distinct associations between DHSs and promoters, CpG islands, conserved elements, and transcription factor motif enrichment. Motif analysis within clusters identified known and novel motifs in cell-type-specific and ubiquitous regulatory elements and supports a role for AP-1 regulating open chromatin. We developed a classifier that accurately predicts cell-type lineage based on only 43 DHSs and evaluated the tissue of origin for cancer cell types. A similar classifier identified three sex-specific loci on the X chromosome, including the XIST lincRNA locus. By correlating DNase I signal and gene expression, we predicted regulated genes for more than 500K DHSs. Finally, we introduce a web resource to enable researchers to use these results to explore these regulatory patterns and better understand how expression is modulated within and across human cell types.
Transcriptional regulation involves a complex interplay of transcription factors (TFs) binding to DNA regulatory elements to control gene expression. This interplay enables a single genome to give rise to hundreds of cell types. Understanding transcriptional regulation requires a full accounting of regulatory elements, including (1) their genomic locations, (2) their cell-type specificity, (3) the identity of factors that bind them, and (4) the genes they target. Ultimately, this accounting will enable us to determine how regulatory elements affect tissue-specific gene expression. In this study, we begin to address these issues by integrating chromatin accessibility and expression data from many human cell types.
Regulatory elements can be identified using chromatin immunoprecipitation (ChIP) experiments, but ChIP requires an individual experiment for each factor and is limited to known factors with previously derived antibodies. Alternatively, regulatory elements can be located TF-agnostically by mapping DNase I hypersensitivity sites (DHSs). DHSs indicate open or accessible chromatin where DNA is not tightly wrapped within a nucleosome, leaving the sequence accessible to DNA-binding proteins (Wu 1980). Genome-wide DNase-seq experiments capture a snapshot of regulatory element dynamics across the multidimensional landscape of cell types, environmental exposures, and developmental stages. Recently, the ENCODE project has made substantial progress defining elements by generating DNase-seq data from more than 100 human cell types (Thurman et al. 2012). Here, we used this extensive collection to provide new insights into tissue-specific regulatory programs. We clustered more than 2 million DHSs from 112 diverse biological samples by tissue specificity into 1856 chromatin profiles and found each cluster to have a distinct bias relative to location, evolutionary conservation, CpG islands, and promoter proximity (distal vs. proximal).
Gene expression profiling has emerged as a powerful tool to classify tumors (Wu et al. 2010). The added resolution of regulatory information may provide a more robust way to classify cell types. To test this, we assigned the 112 samples into tissue groups and developed classifiers to assign tissue type based on DNase I hypersensitivity patterns across the cell-type groups. Our models predicted tissue type with >80% accuracy in leave-one-out experiments. We used this framework to investigate lineage of cancer cell types with a predictor developed using only 43 individual DHSs. A similar model trained to predict the sex of each sample uncovered a set of sex-specific DHSs surrounding three loci on the X chromosome, one of which includes the XIST locus. These results contribute to our understanding of cancer biology and sex determination and highlight the utility of leveraging DNase-seq data across many cell types.
DNase-seq assays typically identify more than 100,000 active regulatory elements in a single experiment, but unlike ChIP experiments, they do not directly reveal which TFs bind to these elements. Many TFs bind to a specific pattern of DNA bases at TF binding sites (TFBSs), often represented as a motif, which can be learned by detecting overrepresented sequences in regulatory elements. Because DNase-seq data from multiple cell types can predict TF binding (Song et al. 2011), the newly available data enable a thorough analysis of many cell types. After clustering DHSs, we used de novo motif discovery to identify relevant known and novel TF motifs and thus predict active TFs that bind to each regulatory element.
Even after identifying TF binding, a key remaining problem is to associate elements with the target genes they regulate (Heintzman and Ren 2009; Stadhouders et al. 2011). These associations can be determined empirically by using chromatin conformation capture (3C) and derivatives to detect long-range chromatin loops (for review, see Wei and Zhao 2011). Unfortunately, three-dimensional (3D) chromatin information often is locus and cell-type specific, and lacks resolution at the level of individual regulatory elements. In practice, typical analyses link elements to genes using heuristics, most commonly by simply assigning them to the nearest gene. Although this is reasonable, it is not always accurate (Noonan and McCallion 2010). Recent studies have pioneered new mapping methods using correlations between expression and other genomic features to link regulators to genes at greater distances and across gene boundaries (Akalin et al. 2009; Ernst et al. 2011). However, linking gene expression to DNase I signal has not yet been explored. We used correlation between DNase I and matched expression data to identify possible target genes for many regulatory elements.
The DNase I and expression data used in this study are accessible within the UCSC Genome Browser (Rosenbloom et al. 2010). However, the linear nature of genome browsers is not ideal for viewing results of the type we present here, which include clustering, motifs, and networks. For that reason, we created a database and web interface to better visualize our analytical results (http://dnase.genome.duke.edu). Through this resource, users can view DHS chromatin accessibility profiles, locate similar sites, and view enriched motifs and predicted target genes. Resources of this type will enable biologists to synthesize meaningful conclusions from integrated experimental results. These results and resources bring us closer to the goal of explaining how chromatin structure relates to transcriptional regulation across diverse human cell types.
Results
DNase I hypersensitive sites cluster cell types by biological similarity
Genomic locations of 2.7 million DNase I hypersensitive sites (DHSs) from 125 samples were described previously (Thurman et al. 2012). From these data, we selected a subset of 112 samples for which we had both DNase-seq and expression data. The 112 samples represent 72 unique cell types and 15 unique tissue lineages (Supplemental Table S1). Data were generated in one of two laboratories, each using a distinct DNase I protocol. We improved on the previously published open chromatin measurements by accounting for batch affects that grouped the data by laboratory rather than by biological signal (see Methods). We used ComBat (Johnson et al. 2007) to remove these batch effects, after which both the DNase I and expression data clustered according to expected biological relationships (Supplemental Fig. S1).
Using these data, we first investigated DNase-seq signals for common patterns across cell types. Previously, we briefly described an initial self-organizing map (SOM) (Wehrens and Buydens 2007) that clustered DHSs by their profile of hypersensitivity across cell types (Thurman et al. 2012). Here, we improved this clustering by increasing the resolution, introducing a step to merge highly similar clusters, and using the batch-corrected data to redefine SOM clusters; we defined 1856 clusters of DHSs (see Methods). This enabled us to identify subtle patterns in the data more robustly and to group similarly acting sites more accurately.
Each DHS was assigned to the single cluster in the SOM that most closely matched its hypersensitivity profile across cell types (Fig. 1A; Supplemental Tables S2–S4). An overall cluster profile (or average DNase I signal in each cell type) was defined by calculating the average hypersensitivity across the DHSs it contained (Fig. 1B). Throughout this study, we refer to clusters using the cell types with increased signal in this averaged DNase I signal profile. We found that multi-cell-type clusters (those whose DHSs were open in more than one cell type) generally involved cell types with known relationships (e.g., Fig. 1B; Supplemental Fig. S2A). In cases in which clusters grouped cell types without obvious biological similarity, this sharing of DHSs may indicate distant lineage relationships, reuse of regulatory elements, transformation related to cancer progression, or may simply reflect a limit in the resolution of the SOM.
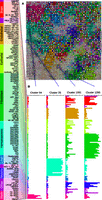
SOM clustering of DHS profiles. (A) A 50 × 50 self-organizing map (SOM). Each box represents a cluster of DHSs with similar DNase-seq signal profiles across samples, color-coded by tissue (legend, left). Cluster color corresponds to the combination of cell types in which the associated DHSs have high signal in the detailed profile. Square size indicates the number of DHSs assigned. (B) Average DHS profiles across samples for four individual clusters. Clusters contain sites open in highly related cell types (54 and 25) and less related cell types (1091 and 1295). (*) Malignant samples.
SOM clusters capture variation in CpG-island, promoter, and conserved element overlap
We annotated each SOM cluster of regulatory elements with respect to overlap with promoters, CpG islands, and evolutionarily conserved elements (see Methods; Supplemental Table S5). We found clear associations between cluster assignment and all three features, which we have illustrated together in a scatterplot (Fig. 2A). For example, clusters in the upper-right corner of the scatterplot (Fig. 2A) are enriched for promoters, CpG islands, and conserved elements, and have a stronger DNase I signal across many cell types (e.g., cluster 99) (Fig. 2B; Supplemental Fig. S2C). Among clusters with similar promoter overlap, the distribution of the distance from DHSs to transcription start site (TSS) varies. For instance, clusters 1361 and 1259 both have 20%–30% promoter overlap, but sites from cluster 1259 are more commonly found just downstream from the TSS (near 5′ introns), whereas sites from cluster 1361 are further from the TSS (Fig. 2B). This finding suggests that DHSs with similar patterns across cell types are likely to share relationships with sequence conservation and genomic location. A striking outlier is the nonpromoter, non-CpG cluster 199, which has an uncharacteristically high conservation score; this cluster, along with other similar clusters, contains ubiquitous distal DHSs that are highly enriched for CTCF motifs (Fig. 2; Supplemental Fig. S2D).
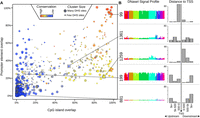
Distribution of conservation, promoters, and CpG islands across clusters. (A) Each cluster is plotted as a bubble. The x-axis indicates the percent of the top 100 DHSs in that cluster (ranked by nearness to the cluster center) that overlap a CpG island; the y-axis indicates the percent that overlap a promoter; color indicates the percent that overlap a phastCons conserved element (Siepel et al. 2005). The size of the bubble indicates the number of DHSs belonging to the cluster. (Red bubbles in the upper-right corner) Clusters capturing primarily highly conserved, CpG-rich promoter elements. (B) DNase I signal profiles of five example clusters, showing the distribution of distance to the transcription start site (TSS) of the nearest gene. Cluster 99 is promoter rich; cluster 1259 is preferentially located in an early intron; cluster 199 is highly conserved, but not associated with promoters or CpG islands; cluster 881 is primarily distal, with no regions within 500 bp of a TSS (see also Supplemental Fig. S2).
A logistic classifier predicts cell-type lineage with few DHS inputs
Since some regulatory elements are highly specific to certain cell types, we reasoned that a subset of elements could be used as molecular markers for identifying cell-type lineage. To test this, we built a multinomial logistic classifier (Fig. 3) that assigns a probability among multiple classes (tissue lineages). Each cell type was first assigned to one of 15 primary tissue types based on known biology (Supplemental Table S1). We removed all malignant cell types and restricted the model to the seven tissue types containing at least four samples each, resulting in a training set of 80 samples across seven classes. Assuming that SOM cluster patterns would be good candidates for differentiating lineages, we used an initial feature set consisting of 1856 DHSs: one from each cluster that was most similar to the average SOM cluster profile. Trained classifiers assigned the highest probability to the correct tissue lineage with >80% accuracy in leave-one-out cross-validation (Supplemental Table S6; see Methods; Supplemental Material). The final model trained using all samples chose only 43 DHSs as informative features (Supplemental Table S7; examples are shown in Fig. 3D). These 43 DHSs are thus one minimum representative set of DHSs with high tissue specificity that can be used to predict tissue identity. The classifier trained using all 80 samples only misclassified two (2.5%) of the 80 samples used to build it: aortic smooth muscle (AoSMC_SFM) and cardiac myocytes (HCM) (Fig. 3A). In these two cases, the model assigned ∼30% probability to the correct lineage (muscle), but a higher (albeit still weak) probability to the fibroblast class. The inability to distinguish between fibroblast and muscle lineages may reflect the biological similarity between them; it is possible to convert fibroblasts into muscle cells in vitro (Tapscott et al. 1988). In addition, regulatory element differences among the included smooth, cardiac, and skeletal muscle samples complicate the muscle lineage and may not be captured by the 43 DHSs used by the model. Samples from blood and stem cells were never misclassified.

Tissue and sex classifiers based on DNase I data. Predictions from a multinomial logistic regression classifier trained to predict tissue identity for a given sample with data from 43 DHSs. (A) Predictions for training data, along with known tissue of origin (left column). Colors within the heatmaps reflect the predicted probability of belonging to each of the seven tissue classes. (B) Predictions for malignant samples not included in the training, but whose presumed tissue of origin was included in the model. (*) Malignant samples. (C) Predictions for samples whose tissue (or presumed tissue) was excluded from the training because tissue types had fewer than five samples. (D) The DNase I signal profiles of seven (out of 43) clusters selected by the model with positive coefficients. (E) The DNase I profile for the single sex-specific site (chrX:130926460–130926610) selected by the classifier. The enlarged barplot shows the distinction between samples divided by sex for the subset of samples included in the model.
To investigate the remaining data, we used this model to classify the 27 malignant samples as well as the five primary cell types left out of the training model (Fig. 3B,C). Fourteen of the malignant samples are presumed to associate with one of the seven lineages that were included in the model (Fig. 3B). For these, the model prediction agreed with this presumed lineage in nine out of 14 cases. Among the five samples whose classifications did not agree, four were derived from brain tumors, and three of these represented specific brain-cell sublineages not present in the training model, which consisted solely of astrocytes. Astrocytes are a subtype of glial cells, which are non-neuronal cells of ectodermal origin. These three brain tumor samples were generally not strongly assigned to any lineage (average maximum probability 34%) (Fig. 3B). The fourth misclassified brain cancer was glioblastoma, which the model confidently (86%) classified as epithelial. Glioblastoma, like astrocytes, originates from glial cells, so this misclassification may indicate differences between astrocytes and other glial cell types, or a substantial remodeling of glial cell chromatin structure that occurs during cancer progression and results in an epithelial-like pattern. In fact, there are reported glioblastoma cases with epithelial differentiation (Rodriguez et al. 2008; Tanaka et al. 2011). This result indicates that this glioblastoma line is more similar to epithelial cell types than to the astrocytes at the chromatin level. The only malignant sample correctly classified as brain was medulloblastoma, which is an embryonal brain cancer consisting of both neuronal and glial cells (Gilbertson and Ellison 2008).
The remaining (nonbrain) misclassification was the K562 leukemia cell line, which we expected would associate with the hematopoietic lineage, but instead weakly associated with multiple lineages, none with probability >30%. The lack of a strong assignment to the hematopoietic lineage may be due to its similarity to undifferentiated erythrocytes (red blood cells), while the hematopoietic lines used to build the model are leukocytes (white blood cells). In contrast, the leukocyte cancer cell types (CLL, CMK, HL-60, Jurkat, and NB4) are all confidently (>75%) assigned to the hematopoietic lineage. This indicates that our blood-specific signatures are not general to all blood cell types, but of the lymphoid lineage only. Another correctly classified sample was Ntera2, a teratocarcinoma cell line often used as a pluripotent stem cell model (Pleasure and Lee 1993), which was appropriately assigned to the stem cell lineage. We similarly evaluated the lineage associations for the remaining excluded samples (Fig. 3C).
We also used SOM-based DHS features to train a predictor to discriminate between male and female samples. We found a single cluster (488) containing sex-specific hypersensitive sites (Fig. 3E). A single representative DHS predicted the correct sex in 40 of 43 (93%) nonmalignant cell types with known sex (Supplemental Table S8). This cluster (488) consists of 30 DHSs on the X chromosome that fall primarily into three loci, one of which surrounds the XIST gene. The second locus includes a noncoding RNA (LOC286467) recently shown to be the only locus on the X chromosome, besides XIST, with sex-specific Pol2 binding (Reddy et al. 2012). The third locus also includes a poorly documented noncoding RNA (LOC550643). Both the second and third loci have complex tandem repeat structures, and all three include annotated piRNAs, which are known to have vital sex-specific roles in germline cells (Girard et al. 2006). Interestingly, these two loci were identified in a recent independent study as having intense H3K4me2 signals on the metaphase X chromosome (Horakova et al. 2012). Each locus was also implicated in inactive-X-specific long-range interactions supporting a role in sex specificity. This result indicates that the SOM method can indeed capture differential regulatory element features in other biological divisions across cell types besides tissue lineage (Fig. 3E).
DHS clusters are enriched for known and novel transcription factor motifs
One motivation for clustering DHSs was to find groups of sites with similar activity profiles, which may indicate commonly bound transcription factors (TFs). We therefore analyzed the clusters for enrichment of TF motifs. We used de novo motif discovery to identify enriched motifs and then assigned motifs to specific factors based on the JASPAR (Portales-Casamar et al. 2010) motif database (see Methods). We found that 1279 (69%) clusters had at least one significant motif (e < 1 × 10−6), while 918 (49%) clusters had a motif that could be assigned a factor from a database (e-value < 1 × 10−6). Alternatively, 1807 significantly enriched motifs were found (some clusters have multiple motifs), of which 1099 (61%) could be assigned a factor (Supplemental Table S9).
We found highly cell-type-specific clusters enriched for motifs known to be important for those cell types, but clusters commonly enriched in a specific cell type did not necessarily share similar motifs, indicating that clusters could discern subtle differences in patterns. Figure 4 provides specific examples of individual clusters and their relevant motif enrichments. For example, the stem-cell-specific cluster 3 was enriched for the known pluripotency factor POU5F1 (Oct-4) motif. The hematopoietic-specific cluster 24 contained the ETV7 (Tel2) motif, consistent with its importance in hematopoietic lineages and leukemia (Potter et al. 2000; Cardone et al. 2005); and an erythroid-specific cluster, 2215, was enriched for GATA family motifs, which are essential for erythroid development (Zhu and Emerson 2002; Ferreira et al. 2005). Interestingly, the motif for the REST repressor was enriched in a medullo-repressed cluster (cluster 36), indicating the potential to also reveal lineage-specific repressive elements. We also found motifs in ubiquitous clusters, discussed further below.
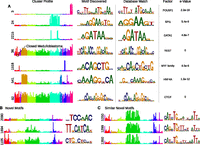
De novo motif discovery results. (A) Representative examples of de novo motif discovery results and highly significant known motif matches. (B,C) De novo motif discovery identified several enriched motifs for which there were no convincing matches to the TF databases. We sometimes found a similar motif across multiple clusters associated with similar cell types.
In 39% of the cases, de novo motifs did not convincingly match known motifs in JASPAR (Fig. 4B), representing possible new or poorly characterized regulators (Supplemental Table S9). For example, in a Urothelium-specific cluster (2090), we identified a short motif (consensus TCCAAC) without a good match in the database. Other clusters (e.g., 1694, 607, 1105, 2142) had similarly high de novo P-values without known motif matches. We found a series of clusters (of which three are depicted in Fig. 4C) that find similar motifs with a CANNTG core sequence and an appendage with ATW consensus 8 bp away. These motifs likely reflect poorly characterized or unknown TFs not yet present in JASPAR, or a complex of TFs.
Motif discovery in similar hematopoietic clusters reveals subtle motif differences
Interferon regulatory factors (IRFs) are DNA-binding proteins that regulate the entire immune response (Paun and Pitha 2007). The DNA-binding domain is highly conserved among the nine human IRF family members (consensus 5′-AANNGAAA-3′), but different IRFs bind slight variations of the core sequence (Fig. 5A; Honda et al. 2006). IRFs may also bind in complex with SPI1, another hematopoietic factor, forming a longer TFBS (Brass et al. 1999). In our analysis, we detected IRF1/IRF2/SPI1-like motifs predominantly in clusters specific to hematopoietic cell lineages, but among these there was variation in DNase I signal intensity among LCLs, B cell leukemia (CLL), T cells (CD4, Jurkat, and Th), megakaryocytes (CMK), and erythroleukemia (K562). We noticed slight variations on the motifs accompanying differences in DNase I signal across hematopoietic cell types (Fig. 5B). This may be due to differences between IRFs and SPI1 binding, different cofactors that modulate an IRF's binding preference, or distinct IRFs in specific hematopoietic lineages. We reason that these motif variations represent biological differences in motif preference rather than statistical noise because in other cases (e.g., in the case of CTCF), we see less variation among discovered motifs across clusters. We also see similar patterns when looking at an independent set of regions from the same clusters (see Supplemental Material; Supplemental Fig. S3A).
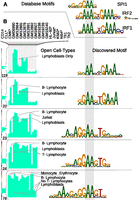
Variations in IRF-like motifs in hematopoietic clusters. (A) Motifs for IRFs and SPI1 from JASPAR show both common and distinct features. (B) MEME motifs discovered in several hematopoietic-specific clusters. The clusters vary in cell-type specificity among the hematopoietic cell types, and the motif logo varies as well, while retaining some semblance of the known SPI1/IRF family motifs.
Motif discovery results are consistent with experimental ChIP data
We used ChIP data from the ENCODE project to validate our discovered motifs (Fig. 6A; Supplemental Table S9). Using representative DHSs from each cluster with enriched motifs (see Methods), we compared overlap with ChIP peaks from 43 experiments (Dunham et al. 2012). We expected some incongruence in overlap between motif and ChIP results because ChIP data come from only a subset of cell types included in the motif analysis. For example, we compared ChIP results for a single IRF from just three cell types, while our motif analysis considered 14 hematopoietic lineages. Without ChIP data for all cell types, we expect to find many instances of a positive motif result without a corresponding ChIP signal. Additionally, ChIP reports signal at indirectly bound sites where a motif would not. Despite these limitations, there is good correspondence (Mann-Whitney P-values between 10−5 and 10−133) between motif enrichment and ChIP results. The correspondence is particularly high for CTCF (Fig. 6A), which is probably due to its cross-cell-type consistency; DHSs in clusters with CTCF motif enrichment and CTCF sites based on ChIP experiments have 96% median overlap, compared with 4% overlap with other clusters. A similar trend is seen for other factors tested. There is high overlap among the IRFs, SPI1, and RUNX1 ChIP and motif results, consistent with all three factors coregulating hematopoietic lineages (Huang et al. 2008). The SP1-motif clusters overlap not only SP1 ChIP peaks, but also ChIP peaks for most of the other factors, consistent with the role of SP1 as a general, promoter-enriched factor with many interacting partners (Kaczynski et al. 2003).
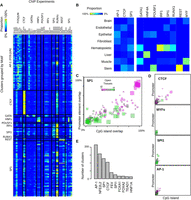
Motif specificity in SOM clusters. (A) Concordance ([yellow] high, [blue] low) between ChIP results (x-axis) and motif discovery in DNase I clusters (y-axis). (B) The cell-type specificity for selected motifs. This heatmap shows the distribution of most-open tissues for each motif. For example, 100% of the clusters where the POU5F1 motif was found had stem cells (Stem) as the most open tissue type, whereas MYF family motifs were found predominantly in muscle clusters. (C,D) Each colored square represents a cluster with enrichment for the given motif. (x-axis) overlap with CpG islands; (y-axis) overlap with promoters; (color) the number of tissues with at least one sample above a cutoff. Each factor shown here has a different distribution of cell-type specificity and promoter/CpG-island overlap. The size of a square indicates the number of DHSs in the cluster. (E) Number of clusters that are enriched for the most common motifs.
Global transcription factor trends suggest AP-1 is a chromatin-accessibility factor
We wanted to know whether individual TFs whose motifs are present in several clusters revealed biologically interesting properties about their function (Fig. 6B; Supplemental Fig. S3). For each TF, we summarized motif results from all clusters and identified lineage trends. We found that TFs with roles in certain cell types were most often enriched in clusters with a small number of relevant tissue lineages. For example, the myogenic factor (MYF) family motif was enriched primarily in muscle-specific clusters, HNF4 in liver clusters, POU5F1 in stem cell clusters, and SPI1 in hematopoietic clusters (Fig. 6B); these are all biologically relevant enrichments (Scott et al. 1994; Nichols et al. 1998; Odom et al. 2004; Cao et al. 2006). This starkly contrasted with ubiquitously expressed transcription factors SP1, AP-1, and CTCF, which did not have a bias toward a single lineage (Fig. 6B). We examined the CpG-content, genomic location, and tissue specificity of clusters where each TF motif was enriched to characterize the regulatory elements that bind each factor. For example, SP1 was enriched in clusters with CpG-island promoters that are present in many cell types (Fig. 6C); this may partly reflect the GC-rich SP1 motif. CTCF was enriched in clusters representing distal DHSs present in many cell types, which is consistent with previous reports (Fig. 6D; Xi et al. 2007; Lee et al. 2012). In fact, we found that the CTCF motif was enriched in all 12 nonpromoter, highly conserved clusters (Supplemental Fig. S2D). The absence of another motif with this property reinforces the uniqueness of function of the CTCF protein. SPI1, MYF family, and IRF family motifs were preferentially enriched in cell-type-specific distal clusters (Fig. 6D). Plots similar to those in Figure 6C were generated for each TF in JASPAR (Supplemental Material).
The most commonly enriched motif discovered was that of Activating Protein 1 (AP-1), found in ∼12% (220) of the clusters. By comparison, the second most common motif, for SP1, was found in ∼8% of clusters (152 clusters) (Fig. 6E). AP-1 is the well-studied FOS:JUN heterodimer that activates both basal and inducible expression (Angel and Karin 1991). It has been implicated in a variety of cellular functions, including cell proliferation, immunity, apoptosis, and differentiation (Angel and Hess 2012). We found the AP-1 motif enriched exclusively in nonpromoter, non-CpG-island clusters (Fig. 6D). In contrast to the tissue-specific factors like MYF family members and SPI1, AP-1 was found in both tissue-specific clusters as well as those shared among many cell types. As detailed in the Discussion, these results suggest that AP-1 may play a general role in chromatin accessibility in many different tissues and genomic locations.
Chromatin and expression signal correlation corresponds with known long-range interactions
The DNase-seq experiment naturally leads to the question of identifying target genes for DHSs. Song et al. (2011) used cross-cell-type correlation among DHSs to identify blocks of similar regulatory elements and coexpressed genes. Thurman et al. (2012) approached this by correlating distal DHSs with promoter DHSs. We reasoned that if the pattern of a DNase-seq signal across cell types matched the pattern of expression of a gene across cell types, this provided evidence that the gene is a regulatory target of the DHS. Therefore, we correlated DNase I hypersensitivity with gene expression data to infer the target genes (both protein-coding and RNA) for each of the ∼2.7 million DHSs (see Methods). About 530,000 of the 2.7 million sites (20%) correlated significantly with at least one gene within 100 kb (permutation P-value < 0.05), a significant enrichment over the 5% expected by chance (Supplemental Table S10). Of these, 71% correlate with a single gene, but some correlate with as many as 44 genes (Supplemental Fig. S4A). 31,000 Ensembl genes (98%) correlated with at least one DHS, and the median number of DHSs associated to a gene was 19 (Supplemental Fig. S4B). Protein-coding genes tended to have more associations than RNA genes (Supplemental Fig. S4C). Figure 7, A and B, illustrates representative examples showing correlations of DHSs to genes that are color-coded to indicate the tissue types that are driving the correlation with gene expression (see Methods). These examples show that associated DHSs can be very far away, crossing multiple gene boundaries.
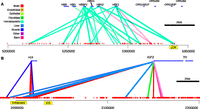
Correlation between DHS and expression. (A) Tie-plot showing the top 50 connections at the beta-globin locus, color coded by tissue type. Red marks below indicate DHSs. Blue bars above represent genes. Connecting lines represent significant correlations, where the width of the lines is proportional to the correlation strength. To simplify the illustration, connections to the olfactory receptors have been removed (see Supplemental Material). (B) Tie-plot for the H19/IGF2 locus (see also Supplemental Fig. S4D).
Long-range regulatory interactions have been previously reported based on chromosome conformation capture (3C) experiments (e.g., Tolhuis et al. 2002). 3C data are not a perfect comparison for several reasons (see the Supplemental Material). Despite these limitations, we compared our results to 3C data and found the 3C and correlation results corroborate one another in eight of 12 cases we investigated (Supplemental Table S11). Two of these are discussed below, with the others described in Supplemental Table S11.
Beta-globin locus
The beta-globin locus control region (LCR) is a collection of five well-characterized DHSs upstream of the beta-globin genes (Molete et al. 2002). The LCR is located near the epsilon-globin gene and has been shown to regulate the other globin genes (HBB/HBD) ∼30–50 kb away in erythrocyte but not in brain cell lineages (Tolhuis et al. 2002). The globin genes are expressed in erythroid cells at different times in development; for example, HBE1 is embryonic, HBG1 and HBG2 are fetal, while HBD and HBB are adult forms (Molete et al. 2002). Our study is limited to detecting connections by the cell types we characterized, and the primary cell type driving connections at this locus is K562 (representing undifferentiated erythrocytes), which is known to express the embryonic globin gene HBE1 (Jackson 2003). Previous 3C experiments showed erythroid-specific proximity between beta-globin genes and the LCR located ∼40 kb from the HBE1 gene (Tolhuis et al. 2002; Palstra et al. 2003). Our results reproduced these findings with a K562-driven link between HBE1 and several downstream hypersensitive sites (Fig. 7A). Most notable, a highly significant correlation linked the beta-globin gene to one of the DHSs in the LCR, hypersensitive site 4 (HS4). This provides a specific association between a particular gene and a particular DHS within the LCR. In addition, there were several other hematopoietic-driven (cyan-colored) links throughout the region (Fig. 7A).
H19/IGF2 ICR
Another well-studied example is the H19/IGF2 locus, which has been shown to have an imprinted long-range interaction (Leighton et al. 1995). In this original study, a 6.2-kb deletion affected expression differently when inherited maternally versus paternally, but this study did not identify individual DHSs that may be involved. An imprinted control region (ICR) located between H19 and IGF2 binds CTCF on the maternal but not paternal allele. When CTCF binds, an enhancer located on the other side of H19 is unable to interact with the IGF2 gene and instead only enhances H19 expression. On the paternal allele, the ICR is methylated, which blocks CTCF binding and allows the enhancer to bind the IGF2 promoter and increase IGF2 expression. While we did not detect interactions with the ICR, we did detect strong correlations between the IGF2 gene and several DHSs located in the H19 enhancer region (Fig. 7B). The correlations were driven primarily by liver lineages, consistent with the role for IGF2 in liver cells. This interaction was detected without any knowledge about allele specificity.
A web resource for exploring DHS sequences, clusters, and TF motifs
The results presented here begin to provide more detailed and informative annotations for 2.7 million DHSs contributing to gene regulation in 112 samples across 72 diverse cell types. To facilitate the further exploration of these data by the research community, we have created a web resource (http://dnase.genome.duke.edu/) to query, display, and extract data. The resource allows queries by regulatory element, by gene, by genome coordinates, by transcription factor, or by cell-type specificity. For researchers starting from a single regulatory element, the web interface provides a list of other regulatory elements with similar cell-type profiles via the SOM clustering. For each SOM cluster, the user can view enriched motifs, genomic distribution, CpG and conserved element overlap, and associated genes and pathways. For any gene of interest, users may view expression, download sets of connected regulatory elements, and explore the clusters to which these connected elements belong. The web resource also enables data to be downloaded in text format for input into genome browsers or external computational pipelines.
Discussion
Our global clustering of DHSs revealed novel open-chromatin pattern relationships among a diverse set of human cell types. Many clusters grouped cell types of common lineage, enabling accurate lineage classifications based on only a few DHSs. We also identified several biologically relevant pathway enrichments for genes near particular clusters (see the Supplemental Material). In future work, we could further delineate among clusters by adding ChIP data for TFs or histone marks, DNA methylation, or DNase I footprinting (Hesselberth et al. 2009; Boyle et al. 2011; Pique-Regi et al. 2011). Creating clusters from a larger set of cell types and developmental stages along with epigenetic data could be a powerful way to characterize cell-type lineage.
The primary experimental data for cell-type-specific TF binding come from ChIP of TFs known in advance (Dunham et al. 2012). We showed that characterizing hypersensitivity across cell types also yields convincing de novo motif discovery results, including identifying novel regulators and new roles for known regulators. This approach provides an unbiased (no a priori knowledge/antibodies required) complement to ChIP, and motifs we discovered in more than 1000 clusters provide a rich resource for further investigation. Our results invite followup study into the function of AP-1, and motifs not yet found in the databases. This resource will also be useful for motif scanning to narrow results based on DHS profiles.
In our motif analysis, the AP-1 motif was the most commonly detected, both in ubiquitous and cell-type-specific clusters of DHSs. Since its subunits (FOS and JUN) are ubiquitously expressed, the cell-type specificity is probably conferred by other factors. This is consistent with a role for AP-1 as a pioneer factor that opens DNA for other factors, or it may be an otherwise general and universal chromatin-accessibility factor. This hypothesis is consistent with experimental results confirming a role for AP-1 in diverse pathways (Angel and Hess 2012). Recent evidence also corroborates the general role of AP-1 in forming accessible chromatin; for example, Shibata et al. (2012) found AP-1 motifs to be associated with chromatin accessibility differences among primates. Similarly, Biddie et al. (2011) showed that inhibiting AP-1 impedes formation of accessible chromatin and reduces glucocorticoid receptor (GR) binding, suggesting that AP-1 has a role in transcriptional pathways mediated by GR. There is also evidence in neurons that AP-1 functions as a general chromatin-accessibility factor, with tissue specificity conferred by cofactors or post-translational modification (Angel and Karin 1991; Weber and Skene 1998). These results are consistent with our finding, which further suggests that this role for AP-1 may be even more general.
Our motif results also highlighted the uniqueness and prominence of CTCF. It is well known that CTCF is an extremely conserved and important factor (Phillips and Corces 2009). Consistent with this, we found the CTCF motif highly significantly enriched in all 12 highly conserved clusters with low promoter overlap (Supplemental Fig. S2). These clusters typically had extreme motif discovery e-values, with >90% of the sequences containing the motif.
Using correlations between DNase I signal and gene expression levels, we predicted mappings between greater than 500 thousand potential regulatory elements and their target genes. We showed that correlation results were often supported by 3C results where these data were available. However, the agreement was not perfect, which is understandable (Supplemental Material): Most importantly, this may be due to either looping interactions or individual DHSs creating poised states without actually affecting expression (Margaritis and Holstege 2008). Nevertheless, open chromatin correlation offers a complement to lower resolution, time-consuming, and expensive chromatin capture-based experiments. This increased level of resolution is necessary for some followup studies, such as increasing resolution of chromatin interaction data, or examining particular SNPs that occur in regulatory elements. Since regulatory mutations likely contribute to complex diseases (Epstein 2009), this type of data will be of clinical interest going forward. By narrowing down vast stretches of noncoding DNA to individual DHSs, we can look for individual SNPs specifically within these sites. As such, DNase I/expression correlation is a powerful additional source of information to inform models of transcriptional regulation.
Methods
Data normalization and processing
See Supplemental Material for the complete Methods. Read data are available at the Sequence Read Archive (Duke: SRX100886-SRX100920 and SRX189386-SRX189433; UW: SRX191006-SRX191058 and SRX201249-SRX201305). DHSs from all samples were combined as described previously (Thurman et al. 2012). For each cell type, we counted the number of DNase I cuts in each DHS. Counts were quantile-normalized and scaled, and protocol batch effects were corrected using ComBat (Supplemental Fig. S1; Johnson et al. 2007).
We used Affymetrix Human Exon 1.0 ST microarrays to measure gene expression. We estimated gene-level expression by normalizing 332 microarray replicates measuring 140 cell lines (data available at GEO; Duke: GSE15805; UW: GSM651582, GSM472913, GSM651582) that included all samples for which we had DNase-seq data (see the Supplemental Material). We combined microarray replicates by taking the median, corrected batch effects, then extracted data for the 112 samples used in this study.
Classifying regulatory elements with a self-organizing map
A self-organizing map (SOM) was constructed using the kohonen R package (Wehrens and Buydens 2007), which was modified to handle more data. Our SOM consisted of a hexagonal 50 × 50 grid (2500 total clusters, or nodes). Since SOMs typically identify many similar clusters, the initially learned SOM was refined by merging similar clusters, resulting in 1856 unique final clusters.
CpG-island, promoter, and conserved element overlap
For each cluster, we extracted the 100 DHSs closest to the cluster center, as assessed by Mahalanobis distance, and tested these for overlap with promoters, CpG-islands, and conserved elements. Promoters were defined as 2 kb upstream of the TSS for the UCSC RefGene annotation (Kent et al. 2002). CpG-island annotations (Bock et al. 2007) and phastCons vertebrate conserved elements (Siepel et al. 2005) were downloaded from the UCSC Genome Browser. We used R bioconductor packages GenomicRanges (P Aboyoun, H Pages, M Lawrence. GenomicRanges: Representation and manipulation of genomic intervals. Bioconductor. http://watson.nci.nih.gov/bioc_mirror/packages/2.10/bioc/html/GenomicRanges.html) and rtracklayer (Lawrence et al. 2009) to do the overlap analyses.
Tissue lineage identity classifier
We used multinomial logistic regression to classify samples by tissue type on the basis of hypersensitivity. Each nonmalignant sample was assigned to one of 15 tissue lineage classes (Supplemental Table S1). Nonmalignant samples from classes with too few (less than five) samples were not used, leaving 80 samples distributed across the remaining classes: brain (five), endothelial (12), epithelial (14), fibroblast (27), hematopoietic (12), muscle (five), and ES (five).
For features, we identified the single hypersensitive site closest to each cluster center based on the Mahalanobis distance. We fit a multinomial logistic regression model using the glmnet R package (Friedman et al. 2010) with leave-one-out (79-fold) cross-validation. We used misclassification frequency as the distance model and used LASSO regularization (alpha = 1) for sparcity. We chose the lambda (regularization) parameter that minimized the misclassification error during cross-validation. Classifications for the malignant cell types were predicted using a model trained with data from all 80 cell types. For the sex classifier, we used a similar model, after filtering malignant samples and those with unknown sex.
Motif analysis
We selected the 100 DHSs from each cluster that were nearest the cluster center, as assessed by Mahalanobis distance. We extracted sequences for these regions and searched them for motifs using MEME (Bailey and Elkan 1994) with the following settings: zero or one occurrence per sequence (ZOOPS), a motif size range of 8–22 nt, and an e-value cutoff of 3 (Supplemental Table S9). After identifying motifs, we used the Bioconductor package motIV (Mercier et al. 2011) to compare the discovered motifs to the JASPAR (Portales-Casamar et al. 2010) motif database, recording the top five matches in each case (Supplemental Table S6).
Mapping regulatory elements to the target genes they regulate
We calculated the Pearson correlation across samples between gene expression and normalized DNase I scores for each DHS within 100 kb of each gene. To reduce noise, we set a minimum value for DNase I signal (0.1) and for gene expression (4). We calculated a permutation P-value by calculating a null distribution of DHS correlations for each gene to a random sample of 10,000 DHSs from different chromosomes, and considered P < 0.05 significant.
Data access
Processed data are available at http://dnase.genome.duke.edu.
Acknowledgments
This work was funded by NIH grants HG004592 (J.A.S.) and HG004563 (G.E.C.), and the University of North Carolina Cancer Research Fund (T.S.F.). N.C.S. was supported by a National Science Foundation Graduate Research Fellowship and the Research Council of Norway.
Footnotes
-
↵8 Corresponding authors
E-mail b.lenhard{at}csc.mrc.ac.uk
E-mail greg.crawford{at}duke.edu
E-mail tsfurey{at}email.unc.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.152140.112.
Freely available online through the Genome Research Open Access option.
- Received November 17, 2012.
- Accepted March 7, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








