
Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms
- Ankit Malhotra1,
- Michael Lindberg1,
- Gregory G. Faust1,2,
- Mitchell L. Leibowitz1,
- Royden A. Clark1,
- Ryan M. Layer1,2,
- Aaron R. Quinlan1,3,4,5 and
- Ira M. Hall1,3,5
- 1Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, Virginia 22903, USA;
- 2Department of Computer Science, University of Virginia, Charlottesville, Virginia 22903, USA;
- 3Center for Public Health Genomics, University of Virginia, Charlottesville, Virginia 22908, USA;
- 4Department of Public Health Sciences, University of Virginia, Charlottesville, Virginia 22908, USA
Abstract
Tumor genomes are generally thought to evolve through a gradual accumulation of mutations, but the observation that extraordinarily complex rearrangements can arise through single mutational events suggests that evolution may be accelerated by punctuated changes in genome architecture. To assess the prevalence and origins of complex genomic rearrangements (CGRs), we mapped 6179 somatic structural variation breakpoints in 64 cancer genomes from seven tumor types and screened for clusters of three or more interconnected breakpoints. We find that complex breakpoint clusters are extremely common: 154 clusters comprise 25% of all somatic breakpoints, and 75% of tumors exhibit at least one complex cluster. Based on copy number state profiling, 63% of breakpoint clusters are consistent with being CGRs that arose through a single mutational event. CGRs have diverse architectures including focal breakpoint clusters, large-scale rearrangements joining clusters from one or more chromosomes, and staggeringly complex chromothripsis events. Notably, chromothripsis has a significantly higher incidence in glioblastoma samples (39%) relative to other tumor types (9%). Chromothripsis breakpoints also show significantly elevated intra-tumor allele frequencies relative to simple SVs, which indicates that they arise early during tumorigenesis or confer selective advantage. Finally, assembly and analysis of 4002 somatic and 6982 germline breakpoint sequences reveal that somatic breakpoints show significantly less microhomology and fewer templated insertions than germline breakpoints, and this effect is stronger at CGRs than at simple variants. These results are inconsistent with replication-based models of CGR genesis and strongly argue that nonhomologous repair of concurrently arising DNA double-strand breaks is the predominant mechanism underlying complex cancer genome rearrangements.
Spontaneous genomic rearrangements are a major source of genetic diversity in cancer and the cause of numerous human disorders. While most genome structural variants (SVs) can be readily categorized into the canonical forms—deletion, duplication, inversion, and translocation—there is growing evidence that a nontrivial fraction are complex genomic rearrangements (CGRs) composed of multiple clustered breakpoints that cannot be explained by a single DNA end-joining or recombination event (Quinlan and Hall 2012).
The existence of CGRs is a very old observation in both the human genetics and cancer fields. Over the years, at least 251 complex rearrangements have been cytogenetically defined in patients suffering from sporadic human disorders (Zhang et al. 2009a), and innumerable complex karyotypic configurations have been reported in human tumors (Mitelman 1994), albeit generally at very low resolution. There are also reports of complex cancer gene amplification events including multifocal clusters (for review, see Albertson 2006), highly rearranged “amplisomes” (Raphael and Pevzner 2004), and chromosome-limited “firestorms” (Hicks et al. 2006).
New, however, is the apparent prevalence of CGRs as revealed by modern genome-wide methods, and the mechanisms put forth to explain them. The initial suggestion that complex SVs might be widespread came from a series of studies characterizing genomic rearrangements associated with sporadic human disorders (Lee et al. 2007; Carvalho et al. 2009; Zhang et al. 2009b). Of 61 nonrecurrent pathogenic mutations, 41% were found to be complex, generally exhibiting multiple adjacent copy number alterations (CNAs) and intra-chromosomal rearrangements. Taking into account previous (for review, see Zhang et al. 2009a) and subsequent studies (Zhang et al. 2010a,b; Choi et al. 2011; Liu et al. 2011a,b; Chiang et al. 2012), these results argue that a large fraction of spontaneous germline mutations are complex in nature. Supporting this, 5%–16% of inherited and presumably benign SVs in mouse (Quinlan et al. 2010) and human (Conrad et al. 2010; Kidd et al. 2010) exhibit multiple clustered breakpoints and/or small-scale insertions or rearrangements at the breakpoint of a larger SV. Complex germline SVs have generally been explained by replication-based models such as fork stalling and template switching (FoSTeS) (Lee et al. 2007), and microhomology-mediated break-induced replication (MMBIR) (Hastings et al. 2009a).
Cancer genome sequencing experiments have revealed highly complex genomic rearrangements involving tens to hundreds of breakpoints that appear to have arisen through a single catastrophic mutational event termed “chromothripsis” (Stephens et al. 2011). The investigators proposed a mechanism involving shattering of large chromosomal regions, perhaps by ionizing radiation or one dramatic cycle of breakage-fusion-bridge, followed by double-strand break (DSB) repair. There is also evidence that chromosome missegregation can generate DSBs (Janssen et al. 2011), and formation of micronuclei at lagging chromosomes can pulverize chromosomes in a manner that might lead to chromothripsis (Crasta et al. 2012). Chromothripsis is likely the same phenomenon as “firestorms,” originally identified in breast cancer array-CGH experiments and found to correlate with patient survival (Hicks et al. 2006).
The true incidence of chromothripsis in cancer, and whether or not different tumor types are more or less susceptible, remain open questions. These questions have been difficult to address because studies have used different methodologies and definitions. Microarray-based estimates of chromothripsis range from 2% to 3% in a diverse set of 746 cancers (Stephens et al. 2011), 1.3% of 764 multiple myelomas (Magrangeas et al. 2011), and 13% of 98 medulloblastomas (Rausch et al. 2012). However, identification of CGRs from microarray data is problematic, and the first two studies appear to have used subjective definitions of chromothripsis, while the latter used a relatively broad definition (10 CNAs on a single chromosome) and enriched for TP53 mutant tumors. Genome sequencing experiments suggest that the true incidence may be higher, at least in certain tumors: five of 25 bone cancers (20%) (Stephens et al. 2011) and 10 of 87 (11%) neuroblastomas showed chromothripsis (Molenaar et al. 2012). Further clouding the issue, prostate cancer genome sequencing has revealed highly complex chains of balanced rearrangements that do not fall under current definitions of chromothripsis (Berger et al. 2011). Interestingly, the incidence of chromothripsis in medulloblastomas correlates with TP53 loss (Rausch et al. 2012), indicating a potential link to DNA damage response or apoptosis and suggesting that different tumors may have a variable incidence depending on genetic background. However, the relationship is likely to be more complicated since no association between genic mutations and chromothripsis was detected in neuroblastomas despite whole-genome mutation data (Molenaar et al. 2012).
The human genetics and cancer fields have converged with the description of chromothripsis events in the germline that closely resemble those reported in cancer cells (Kloosterman et al. 2011a, 2012; Chiang et al. 2012), and with the proposition that the DNA replication-based mechanisms originally proposed to explain relatively mild germline CGRs may also underlie chromothripsis in cancer (Liu et al. 2011b). This raises the important question of whether or not complex mutations in germline and somatic cells have a common origin. It has been difficult to address this question because most events characterized thus far in germline lineages are relatively mild, presumably due to ascertainment bias related to selective pressures acting during early development, and because cancer genome studies have thus far focused on the most complex subset of events.
Here, we perform a systematic screen for CGRs in 64 tumor genomes, use de novo assembly to profile rearrangement breakpoints at single-base resolution, and compare mutational signatures and intra-tumor allele frequencies at both simple and complex mutational events.
Results
Breakpoint mapping
This study includes 64 tumors from The Cancer Genome Atlas (TCGA) including 12 basal-like breast cancers (BRCA), three colon adenocarcinomas (COAD), 18 glioblastomas (GBM), six lung adenocarcinomas (LUAD), 13 lung squamous cell carcinomas (LUSC), 11 ovarian cancers (OV), and two renal adenomas (READ) (Supplemental Table 1). Tumor and matched normal samples, in the form of blood or normal solid tissue (in one case, both), were subjected to Illumina paired-end sequencing by TCGA.
To identify SV breakpoints, we used HYDRA-MULTI, a new multisample version of our HYDRA paired-end mapping algorithm (Quinlan et al. 2010) that uses population-based clustering (Quinlan et al. 2011). Read pairs from all 64 tumor samples and 65 normal samples were combined into a single clustering step, which enabled simultaneous measurement of the evidence for each breakpoint in each sample. This method and several filtering steps (see Methods) identified 6179 somatic rearrangement breakpoints. For simplicity, SV breakpoints with distance <1 Mb are classified as deletions, tandem duplications, or inversions based purely on their orientation. The remaining breakpoints are classified as either “inter-chromosomal“ or “intra-chromosomal.” We note that this classification may not necessarily reflect variant type; for example, inverted duplications can produce apparent “inversion” breakpoints, and both inversion and inter-chromosomal breakpoints are often associated with CNAs (Supplemental Fig. 4). Different tumors and tumor types show different numbers and types of breakpoints (Fig. 1A), as reported previously (Stratton 2011), with BRCA and LUSC samples often showing large numbers of tandem duplications, and GBM samples showing numerous large-scale rearrangements. We also identified 27,093 germline breakpoints, of which we use a high-confidence set of 9964 deletions and 1980 tandem duplications as controls in subsequent analyses.
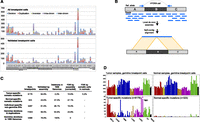
HYDRA-MULTI breakpoint calls. (A) Stacked bar graph displaying the number of SVs in each tumor, with different SV classes shown as different colors. The top plot has all SV calls; below are the calls validated by assembly. In the legend, deletions, tandem duplications, and inversions are smaller than 1 Mb; (intra-chrom) intra-chromosomal rearrangements larger than 1 Mb; (inter-chrom) inter-chromosomal rearrangements. (B) Assembly-based validation of a breakpoint call corresponding to deletion of the “B” segment. Read pairs in which one or the other read maps near a breakpoint prediction are extracted and subjected to de novo assembly. Contigs are then aligned to the reference genome. Split alignments detecting breakpoints consistent with the original call are judged as validated. (C) Table showing the validation results for different breakpoint callsets. From top to bottom, the rows correspond to somatic mutations predicted in a single tumor sample, “somatic mutations” predicted in a single normal sample, SV calls present in a single tumor–normal pair, germline control breakpoints, and germline deletion calls that were also found by the 1000 Genomes Project. The “Deletions in 1000 Genomes” column shows, for each subset of calls shown in rows, the percentage of deletions that were also found by 1000 Genomes, defined as 50% reciprocal overlap. The last two columns show the FDR by assembly, and by assessing the number of normal specific somatic mutations. (D) The top two panels show the number of germline breakpoint calls found in tumor (left) and normal (right) samples. The bottom panel shows the number of breakpoint calls found in a single tumor sample (left), but no other sample, or a single normal sample (right).
Since DNA was not available, we used local de novo breakpoint assembly to assess the validation rate. We modified the SGA assembler (Simpson and Durbin 2012) to report all paths through the assembly string graph, rather than just a consensus contig. This allows for assembly of breakpoints present at relatively low (<50%) allele frequencies within tumor cell populations, as the vast majority of somatic SVs are. Contigs exhibiting split alignments consistent with the original breakpoint prediction were judged to validate the call (Fig. 1B). Using this method, we validated 64.8% of somatic breakpoints and 58.5% of germline control breakpoints (Fig. 1; Supplemental Tables 8, 9), with a median contig length of 862 bp. However, breakpoint assembly is technically difficult and may fail to produce validating contigs. For example, we were only able to assemble and validate 76.8% of the 5368 deletions that were identified by both our study and the 1000 Genomes Project (Mills et al. 2011), and validated by the latter. Assuming that 100% of the shared calls are true positives, this implies a validation rate of 84.4% for somatic breakpoints, corresponding to a false discovery rate (FDR) of 15.6%. This is likely an overestimate of FDR since in our experience deletion polymorphisms are the easiest SV class to assemble and validate.
As an independent test of accuracy, we assessed the relative number of somatic breakpoint calls in tumor versus normal samples and found that calls private to a single sample are overwhelmingly enriched in tumors (Fig. 1C,D). Of the 6502 breakpoint calls detected exclusively in one of the 129 data sets, 6179 (95%) were observed in a single tumor, whereas a mere 323 (5%) were found in a single normal sample. We expect that most of the 323 normal-specific “somatic mutations” are false positives, although some may result from bona fide somatic mutation or from loss of heterozygosity in tumor samples. Since tumor and normal data sets have similar genomic coverage (Supplemental Table 1) and a similar number of germline breakpoint calls (Fig. 1D), it is reasonable to expect that the false-positive rates would be similar. Thus, notwithstanding a tumor-specific source of false-positive somatic breakpoint calls (of which we have no evidence), these data imply that roughly 323 of the 6179 somatic breakpoints are false calls, yielding an FDR of 5.2%. Given the extremely well controlled nature of this tumor–normal comparison, we believe that this FDR estimate is more accurate than that obtained by breakpoint assembly.
Genotyping errors are a common source of false-positive somatic SV calls in cancer sequencing studies, since a germline breakpoint may be misclassified as somatic due to a false negative in the matched normal sample. Importantly, the calculation outlined above includes this source of errors. Further supporting a low rate of somatic misclassification, only 2.2% of the 1822 somatic deletion calls correspond to known deletions from the 1000 Genomes Project calls, in contrast to 53.9% of the 9964 germline deletion calls (Fig. 1C).
We cannot measure the true false-negative rate, but we can obtain an approximation by assessing the discordance between matched tumor–normal data sets. Of the 131,638 positive genotype calls made for inherited germline SVs in either tumor or normal data sets, 78,945 (59.97%) were made in both matched data sets. This suggests a false-negative rate (FNR) of ∼40% at SV breakpoints captured by HYDRA-MULTI.
A caveat to these analyses is that we excluded small inversions (<10 kb) due to a previously undescribed library preparation artifact in TCGA data that produces numerous false inversion calls in the 1-kb to 10-kb size range (see Methods). Despite this filter, the assembly-based validation rate for inversions (54%) is substantially lower than for large-scale rearrangements (80.3%), tandem duplication (91.8%), and deletions (92.5%), and one sample (BRCA_3_T) is plagued by 113 unvalidated inversion calls in the 10-kb to 20-kb size range (Fig. 1A; Supplemental Tables 8, 9). We have included the 588 inversion breakpoints in subsequent analyses due to their utility in helping to define the architecture of certain complex rearrangements, but we note that removing them does not significantly alter our findings or conclusions.
Complex breakpoint clusters are common in cancer genomes
Visual inspection of somatic breakpoint calls revealed complex breakpoint patterns in many tumor genomes, including dense clusters of adjacent and/or intertwined breakpoints and chains of interconnected rearrangements. To systematically identify sets of three or more interconnected breakpoints, which we refer to as “breakpoint clusters” or simply “clusters,” we developed a method involving two steps (Fig. 2A): (1) define breakpoint loci by merging calls whose mapping positions in the reference genome are within 100 kb of one another; and (2) chain together loci that share breakpoint calls in common, which is possible because each breakpoint in the experimentally sequenced cancer genome represents a junction between two distinct loci in the reference genome. In this manner, breakpoint clusters can involve multiple loci in the reference genome that have been rearranged into a single contiguous region of the test genome. The end result is that all the breakpoints in a cluster are interconnected and no farther than 100 kb from another breakpoint in the cluster.
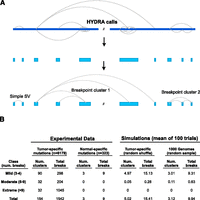
Detecting complex genomic rearrangements (CGRs). (A) HYDRA-MULTI calls are shown as dotted lines connecting distinct loci in the reference genome (blue bar at top), with each call predicting a single novel junction in the test genome corresponding to exactly two loci in the reference. Breakpoints found within 100 kb of each other are merged, and “breakpoint clusters” are formed by chaining together loci linked by one or more breakpoint calls. (B) Table showing the results of breakpoint clustering and simulation, broken down by severity (as defined at left). The left half of the table shows breakpoint clusters identified from experimental data, using either tumor-specific somatic mutations or normal-specific “somatic mutations” (false positives). The right half shows simulation results based on randomly shuffling genomic coordinates of somatic SVs, or from randomly sampling 1000 Genomes deletions.
We retained clusters involving three or more distinct breakpoint calls. To minimize fragmentation, where subsections of the same apparent rearrangement may be reported separately due to false-negative breakpoint calls, we merged nearby clusters using a distance threshold of 1 Mb. These methods identified 154 breakpoint clusters involving 1542 of the 6179 (25%) total breakpoints (Fig. 2B). Of these, 90 were “mild” clusters composed of three to four breakpoints, 32 were “moderate” (five to nine breakpoints), and 32 were “extreme” (10 or more breakpoints). Although we used a clustering threshold of 100 kb, most breakpoint clusters are remarkably dense. The median inter-breakpoint distance within clusters is 1.5 kb (compared to 545 kb for all somatic breakpoints), 74.5% of breakpoints are within 10 kb of another breakpoint in the cluster, and 43.1% are within 1 kb (Supplemental Fig. 1). Breakpoint clusters were identified in 48 of 64 genomes (75%) representing all seven cancer types and are relatively evenly distributed across tumor types, with most tumors showing one to five clusters (Fig. 3D). Thus, complex patterns of genomic rearrangements are detectable in most cancer genomes.
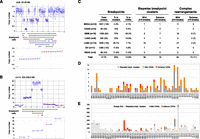
Prevalence of CGRs. (A) An example of copy number state determination. On top is a plot showing the raw (blue dots) and segmented (red line) read-depth data at a CGR with three copy number states. (Middle panel) HYDRA-MULTI breakpoint calls, with deletions (red), tandem duplications (green), and inversions (blue). The bottom panel shows the results of clustering CNA change-points into copy number states, where copy number values are sorted in ascending order and clusters with the same state are shown with a blue line. (B) Same as panel A, except a stepwise rearrangement with five copy number states. (C) The number of stepwise and complex rearrangements by tumor type, as shown at left. Columns correspond to the total number of breakpoints, the percent that are in clusters, the percent judged to be complex, and the number of mild and extreme events for stepwise and complex rearrangements. For the rightmost column, the number of samples exhibiting extreme CGRs is shown in parentheses. (D) The number of CGRs observed for each tumor. (E) The number of breakpoints in each tumor, broken down by complexity class.
To evaluate the significance of these results, we performed a variety of control experiments (Fig. 2B; Supplemental Fig. 2). We conducted a Monte Carlo simulation by shuffling breakpoint positions within uniquely mappable regions of the reference genome, controlling for the size distribution and class of SV calls, and discovered a mean of 4.9 breakpoint clusters per iteration. This is in stark contrast to the 154 found among all the samples in the real data. To control for the potential effect of false-positive breakpoint calls, we applied our method to “somatic” breakpoints private to a single normal sample and identified merely three clusters. To control for the nonrandom distribution of germline SVs, we performed simulation experiments by randomly sampling breakpoints from three high-confidence callsets: validated 1000 Genomes Project deletion calls, validated germline breakpoints from this study, and “rare” germline SVs identified in a single tumor–normal pair. These experiments yielded a mean of 3.1, 11.2, and 12.6 breakpoint clusters, respectively, the vast majority of which had three to four breakpoints. These results demonstrate that only a very small number of breakpoint clusters are identified by chance and that clusters identified by chance have very few breakpoints.
Consistent with our simulations, breakpoint clusters are not enriched at known SV hotspots such as segmental duplications or common fragile sites, nor at repetitive elements known to produce spurious SV calls due to read mapping artifacts (Supplemental Fig. 3).
On the origin of breakpoint clusters: Single versus multiple mutational events?
A breakpoint cluster may result from a complex one-off mutational event that simultaneously generates multiple breakpoints, or from a series of simple mutations that occur in stepwise fashion. Although our simulations clearly demonstrate that breakpoint clusters are very rarely identified by chance under a model of random mutation, tumor genomes do not necessarily evolve through random processes. Breakpoint clusters may be generated by breakage-fusion-bridge cycles that promote repeated rounds of mutation within a chromosome arm, or from progressive amplification of genes that confer fitness advantage. There is no foolproof method to distinguish between one-off and stepwise mutations, and thus there is no definitive way to prove the mutational origin of any given breakpoint cluster. An informative feature for inferring the most likely mutational scenario is the number of DNA copy number states associated with a breakpoint cluster. One-off mutations caused by repair of multiple DNA breaks have limited ability to generate multiple copy number states because DNA breakage and ligation can only involve the small number of chromosomes inside of a cell at any given time, and most reported chromothripsis events involve three or fewer states (e.g., loss, gain, and unaltered) (Kloosterman et al. 2011b; Magrangeas et al. 2011; Stephens et al. 2011; Molenaar et al. 2012; Rausch et al. 2012). Replication-based mechanisms such as MMBIR can in theory generate an unlimited number of states in a one-off event, and there have been reports of triplication (Carvalho et al. 2011; Liu et al. 2011b), but to our knowledge most if not all variants attributed to MMBIR also exhibit three or fewer states. In contrast, stepwise mutations often produce numerous copy number states due to the likelihood that new CNAs arise within older CNAs.
To detect CNAs, we performed circular binary segmentation (Olshen et al. 2004) of GC-normalized read depth measured in windows containing 5 kb of uniquely mappable sequence (Quinlan et al. 2010). We refer to the junctions between adjacent genomic segments with distinct copy number as “change-points.” Supporting the quality of our CNA callset, ∼38% of all somatic breakpoint calls and 63% of large (>100 kb) duplication and deletion calls are within 10 kb of a CNA change-point, which represents a 47-fold and 139-fold enrichment based on simulations, respectively (Supplemental Table 7). To accommodate the imprecision of read-depth analysis and to ensure that CNA states were not underestimated, change-points within 100 kb of a breakpoint cluster were included in CNA state analyses. Of the 154 clusters, 83.8% are associated with at least one CNA, and 45.5% with at least 10 CNAs (Supplemental Fig. 4). We then used a custom algorithm (see Methods) to estimate the number of CNA states at each breakpoint cluster (Fig. 3A,B). To ensure accuracy, we evaluated all CNA state determinations by eye (Supplemental Figs. 6–8), visualized breakpoint clusters using Circos (Krzywinski et al. 2009) and IGV (Robinson et al. 2011), and selected conservative parameters to minimize misclassification of stepwise rearrangements.
For a breakpoint cluster to be judged as a complex rearrangement resulting from a one-off mutation, we required that it exhibited no more than three copy number states and no more than one amplified copy number state exceeding four predicted copies, and that it was not a focal amplification composed of a single contiguous amplified region. These criteria are consistent with previous studies of chromothripsis and arguably more precise. A caveat is that although this method reveals many crystal-clear examples of stepwise (Fig. 4) and one-off events (Figs. 5, 6), false-negative variant calls sometimes cause apparent misclassification, and there are boundary cases that might be classified differently under distinct rules or parameters. It is also important to recognize that stepwise and one-off mutational processes are not mutually exclusive and that some breakpoint clusters may result from a combination of both.
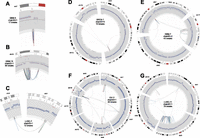
Circos plots of stepwise breakpoint clusters. Only the chromosome(s) and breakpoints involved in the rearrangement are shown. Chromosome coordinates increase in the clockwise direction. The chromosome name is indicated outside the circle. The outermost track is the cytogenetic band, with the centromeres (red). Moving inward, the second track is COSMIC cancer genes. Next is a plot showing the copy number profile obtained from read-depth analysis. This profile includes germline CNVs and somatic CNAs. The track shows normalized read depth, represented as a Z-score (blue dots) and segmented read-depth data (red line plotted on top of the blue dots). The y-axis limits correspond to the median Z-score ±7.5 median absolute deviations. The next track shows the somatic CNA change-points (lighter gray track inside of the read-depth track). Rearrangements are depicted as lines connecting points on the circular chromosome(s) with deletion breakpoints (red), duplications (green), and inversions (blue). Note that these breakpoint classes are defined by the relative orientation of the joined genomic segments, and may not actually involve deletion or duplication of sequence. (A) A focal amplification at the EGFR gene. (B) A multifocal amplification. (C) Coamplification linked by inter-chromosomal rearrangement. (D–F) Increasingly complex patterns of amplification plus rearrangement. (G) A CGR from a LUSC genome with a highly rearranged chr3q, perhaps due to breakage-fusion-bridge.
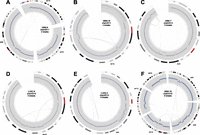
Circos plots of mild CGRs following the conventions outlined for Figure 4.
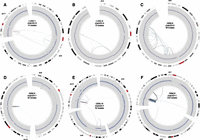
Circos plots of extreme CGRs (chromothripsis) following the conventions outlined for Figure 4.
Using these criteria, 97 of the 154 breakpoint clusters (63%) are consistent with being generated by a one-off mutational event. We hereafter refer to these as complex genomic rearrangements (CGRs). CGRs are found in 43 of 64 tumors (67%) and account for 13.6% of all somatic breakpoints. There are 13 “extreme” CGRs comprising 10 or more breakpoints, but the vast majority of CGRs are relatively “mild” events that would not be apparent using array-based methods (Fig. 3C,D). These analyses indicate that complex one-off mutations play a major role in shaping cancer genome architecture.
High incidence of chromothripsis in glioblastoma
The prevalence of chromothripsis in tumor genomes remains an open question. A complication is that there is no accepted definition of chromothripsis, and previous studies have used different definitions depending on the resolution of the underlying data. Given the availability of whole-genome sequence data for all samples in this study, and the existence of highly complex rearrangements that are mostly, if not entirely, balanced (Supplemental Figs. 5, 6; Berger et al. 2011; Chiang et al. 2012), we prefer a simple and unbiased definition based purely on the presence of 10 or more clustered breakpoints and copy number profiles consistent with one-off mutation (as defined above). Using this definition, there are 13 examples of chromothripsis among 11 tumor genomes (Fig. 6; Supplemental Fig. 9). Remarkably, nine chromothripsis events were found in seven of the 18 GBM samples, and merely four events were found in four of the remaining 46 non-GBM tumors (Fig. 3C,D). This represents an incidence of 38.9% in GBM and 8.7% in non-GBM samples, which is a statistically significant difference by a Fisher's exact test (P = 0.0079). To our knowledge, this is the first demonstration that chromothripsis is a variable phenotype among tumor types. The prevalence of chromothripsis is not correlated with the number of breakpoints detected among tumor types (Fig. 3E). For example, the 12 BRCA genomes have an extremely high SV burden, with a mean of ∼138 breakpoints per tumor, but only 2.1% of breakpoints are in CGRs and there are no examples of chromothripsis. In contrast, the 18 GBM samples have fewer than half the mean number of SV breakpoints (∼60 per tumor), but 49.3% of breakpoints are in CGRs and there are nine examples of chromothripsis. LUSC samples have a high SV burden (mean 139 per tumor) and a high incidence of both stepwise breakpoint clusters and mild one-off CGRs, but only two examples of chromothripsis. These data show that, relative to other tumor types, GBM samples are especially prone to catastrophic genomic rearrangements.
The above definition of chromothripsis is based purely on the number of SV breakpoints and does not require numerous CNA change-points, as in previous microarray-based studies. If we define chromothripsis as extreme CGRs with 10 or more SV breakpoints and 10 or more CNA change-points, there are nine chromothripsis events from six tumors, and all but one event is in a GBM sample. Thus, if we restrict our definition to the most extreme versions of chromothripsis that likely would have been detected by previous studies, the enrichment of chromothripsis events in GBM samples is even stronger and remains significant (Fisher's exact; P = 0.0055).
Finally, the most rigorous definition of chromothripsis relies on performing a Monte Carlo simulation for each putative event to test whether the observed rearrangement breakpoints, applied to an in silico chromosome in stepwise fashion and random order, produce significantly more CNA states than observed in the data (Stephens et al. 2011). We used this simulation strategy to test the 13 extreme CGRs composed of 10 or more breakpoints (see Methods). We find that seven of 13 extreme CGRs have significantly fewer CNA states than expected by chance under a stepwise model (P < 0.05). The seven events are found in seven different tumors, five of which are GBMs, and the increased incidence in GBM samples remains statistically significant (Fisher's exact; P = 0.0157). A caveat to this analysis is that, since the simulation-based statistical test relies on the relationship between breakpoints and experimentally detectable CNA states, it is only suitable for CGRs with numerous intra-chromosomal rearrangements and large CNAs, not those composed primarily of balanced rearrangements. It is also unclear how many chromothripsis events reported in the literature would pass this test, since so far it has only been applied to a small subset of previously reported examples (Stephens et al. 2011; Northcott et al. 2012; Rausch et al. 2012).
Complex breakpoint clusters resulting from stepwise mutation
Breakpoint clusters encompass a broad spectrum of complexity and exhibit vast architectural diversity (Figs. 4–6). Given the difficulty in fully describing this diversity, we encourage the reader to peruse Circos plots (Krzywinski et al. 2009) of the breakpoint calls for each cluster (Supplemental Figs. 5–7), as well as for the entire genome of each tumor (Supplemental Fig. 12). Here, we discuss a few trends.
We define three breakpoint classes: (1) local alterations <1 Mb in size; (2) intra-chromosomal rearrangements >1 Mb; and (3) inter-chromosomal rearrangements. Amplified loci have predicted copy number greater than four.
Of the 57 breakpoint clusters predicted to result from stepwise mutation, six closely resemble one-off mutations but exhibit four copy number states. The other 51 are associated with amplified loci including numerous known cancer genes. Remarkably, only 19.3% are local (Fig. 4A), with the remainder showing intra-chromosomal rearrangements (45.6%), inter-chromosomal rearrangements (10.5%), or both (22.8%) (Fig. 4B–G). Thus, complex patterns of gene amplification are the rule, not the exception. It is unclear whether amplification causes rearrangements or vice versa, or whether both are caused by the same regional genetic instability. A significant fraction of stepwise breakpoint clusters were identified in LUSC genomes (43.9%), and 15 of these are found on highly rearranged chromosome arms potentially resulting from breakage-fusion-bridge (e.g., Fig. 4G). Chr3q is highly rearranged in six of 13 LUSC genomes.
Interestingly, there are 13 stepwise breakpoint clusters in which rearrangements join coamplified regions of the genome (Fig. 4B–F; Supplemental Fig. 11). These “amplisomes” were first noted during studies of the MCF7 breast cancer cell line (Raphael and Pevzner 2004). This suggests that an important consequence of cancer genome rearrangement may be to shuffle genes into configurations that enable coamplification. Several clusters exhibit multifocal amplifications interdigitated with segments of unaltered copy number at a single locus, with rearrangements linking amplified segments (e.g., Fig. 4B). Multifocal amplifications have been observed in array-CGH experiments but were interpreted as independent events (Albertson 2006); our data indicate that they may often be present as a single amplicon. The presence of multiple amplified CNA states in amplisomes argues that an initial amplification often precedes rearrangement, followed by subsequent coamplification of the rearranged genomic segments. Not all coamplified segments contain known cancer genes, as defined by COSMIC (Shepherd et al. 2011), but there are notable examples including MYCL1 and SOX2 (Fig. 4C); MDM2, CDK4, and DDIT3 (Fig. 4B); and PIK3CA, SOX2, and MLF1 (Supplemental Fig. 11, p. 9).
The landscape of complex genomic rearrangement
We now turn our attention to the 97 CGRs predicted to result from a single mutational event (Figs. 5, 6; Supplemental Figs. 5, 6). Only 21.6% are local, with the remainder showing intra-chromosomal rearrangements (40.2%), inter-chromosomal rearrangements (23.7%), or both (14.4%). The majority (74%) are associated with CNAs. Large-scale rearrangements often connect CNAs from distinct genomic regions on one or more chromosomes, focal breakpoint clusters and small CNAs often occur at the edges of larger CNAs, and apparently contiguous CNAs may contain numerous cryptic internal rearrangements involving small segments of unaltered copy number (e.g., see Supplemental Fig. 6, p. 1). Thus, many CNAs that would appear to be derived from independent mutational events by array-CGH or read-depth analysis are, in fact, derived from complex mutations involving both CNAs and rearrangements. Chromothripsis events show remarkably diverse architectures including a single dense breakpoint cluster (e.g., Fig. 6D), multiple dense breakpoint clusters linked by large-scale rearrangement (e.g., Fig. 6E,F), and more diffuse events spanning large chromosomal regions (Fig. 6A–C). Only nine one-off CGRs are associated with amplified loci, and seven of these may be stepwise CGRs misclassified due to false-negative SV calls; however, one resulted in a highly complex MDM2 amplification marked by numerous oscillations between copy number loss and high-level amplification, presumably due to double minute formation via chromothripsis (Fig. 6D).
Finally, inspection of rearrangement patterns across the entire spectrum of CGRs suggests that chromothripsis may be the most extreme manifestation of a common underlying mutational process. It is difficult to quantify this observation, but in a qualitative sense the rearrangement patterns observed among chromothripsis events (Fig. 6) are reiterated among less complex CGRs (Fig. 5). For example, many of the 18 CGRs exhibiting five to nine breakpoints are ostensibly similar to chromothripsis events, with multiple breakpoint clusters linked by larger-scale rearrangements, and multiple CNAs representing two copy number states. The main difference between these CGRs and those attributed to chromothripsis is the number of breaks contained in each cluster, and the number of clusters. However, this difference in CGR severity as related to breakpoint number and density is, in our view, more likely to reflect differences in the severity of DNA damage events provoking rearrangement, not a distinct mechanism per se. Thus, the distinction between chromothripsis and mild CGRs may be one of degree, not of substance, and the majority of one-off CGRs may arise through a common mechanism.
CGRs have elevated intra-tumor allele frequencies
A key question is whether CGRs are generally early events in tumorigenesis or whether they are more commonly late events arising perhaps due to acquired genomic instability in the mature tumor. To address this question, we measured the intra-tumor breakpoint allele frequency (BAF) by aligning raw reads to the junction sequences representing the alternate and reference alleles (see Methods). As expected, application of this method to germline breakpoints produced a BAF centered at 0.5 corresponding to heterozygous SVs, and a peak at 1 corresponding to homozygous SVs (Fig. 7A). In contrast, somatic SVs generally have BAFs lower than 0.5, which is expected given tumor heterogeneity and the presence of stromal cells in most samples. However, there is a subtle yet significant difference between simple SVs and breakpoint clusters. Whereas their mean BAFs are roughly similar (0.34 vs. 0.374), breakpoint clusters have a higher median frequency (0.308 vs. 0.361), and this rightward shift in the distribution is significant by the Mann-Whitney-Wilcoxon (MWW) test (P = 4.19 × 10−9). As such, 53.7% of breakpoints found in clusters have “high” BAFs (>0.35), whereas only 39.8% of simple SV breakpoints do (Fig. 7C). This is not likely to be explained by amplifications, which could artificially elevate allele frequencies at breakpoints within amplicons, because a similar fraction of simple SV breakpoints have unusually high BAF (>0.65) as clustered breakpoints (4.8% vs. 5.0%). Thus, if we only assess breakpoints that appear to arise early during tumorigenesis but not to be affected by CNAs or LOH events, which we define as a frequency of 0.35–0.65, 48.7% of clustered breakpoints but only 35.0% of simple SVs fall within this range.
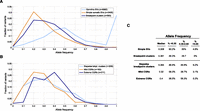
Intra-tumor breakpoint allele frequency (BAF). (A) BAF distribution for germline duplications and deletions (light blue), simple SVs (orange), and complex breakpoint clusters (dark blue). For germline breakpoints present in multiple samples, each BAF measurement of that breakpoint is shown. (B) BAF distribution for stepwise and complex rearrangements. (C) Table showing the median BAF for different breakpoint classes, and the percentage of breakpoints that have low (<0.35), high (0.35–0.65), and unusually high (>0.65) BAF.
If we compare stepwise rearrangements, mild one-off CGRs composed of three to nine breakpoints, and extreme one-off CGRs composed of 10 or more breakpoints (chromothripsis), the difference in BAF between simple SVs and breakpoint clusters is primarily due to chromothripsis (Fig. 7B). Remarkably, 63.5% of chromothripsis breakpoints have BAFs higher than 0.35. Relative to simple SVs, mild CGRs are not significantly different, stepwise breakpoint clusters show a subtle yet mildly significant difference (MWW; P = 0.016), and chromothripsis breakpoints show a large and highly significant difference (MWW; P = 3.34 × 10−13).
These results indicate that highly complex CGRs often arise early during tumorigenesis or, alternatively, are often under strong selection and rise to high frequency. Either scenario implicates complex rearrangements as a functionally important form of tumor genome evolution.
CGRs are predominantly formed by end-joining
The mechanism(s) of CGR formation is an unresolved question. There are two general models: (1) template switching at a DNA replication fork (FoSTeS/MMBIR) (Lee et al. 2007; Hastings et al. 2009a) or bubble (Howarth et al. 2011); and (2) chromothripsis, which involves chromosome shattering followed by nonhomologous or microhomology-mediated end-joining (NHEJ/MMEJ) (Stephens et al. 2011). There is evidence for both models. Sequencing of several hundred chromothripsis breakpoints in cancer genomes (Kloosterman et al. 2011b; Stephens et al. 2011; Rausch et al. 2012) and 282 CGR breakpoints from germline genomes (Kloosterman et al. 2011a, 2012; Chiang et al. 2012) has led some to propose that end-joining is the predominant cause. On the other hand, detailed molecular characterization of CGRs underlying sporadic human disorders has led others to support DNA replication-based models (Lee et al. 2007; Carvalho et al. 2009; Hastings et al. 2009a,b; Zhang et al. 2009a,b; Liu et al. 2011b).
In practice, it is difficult to distinguish between template switching and end-joining because both mechanisms can use stretches of microhomology (2–10 bp) and both can lead to small-scale DNA insertions or rearrangements at the breakpoint. Moreover, despite the elegance of MMBIR for explaining certain CGR architectures (e.g., triplication), end-joining can, in principle, lead to any conceivable CGR architecture given a sufficient number of chromosomes and breaks. However, MMBIR has one strict requirement that end-joining does not: It requires microhomology. To our knowledge, no DNA polymerase can initiate template-directed synthesis without a primer.
We therefore profiled homology at SV breakpoints by measuring “alignment overlap” at split-contig mappings (Fig. 8A). When the entire distribution is considered, germline and somatic breakpoints show very different profiles (Fig. 8B). First, there are numerous germline breakpoints with 10–20 bp of homology. These correspond to inherited LINE and SINE insertions present in the reference genome but not one or more test genomes; apparent homology results from target site duplications. Second, exceedingly few somatic breakpoints are formed by nonallelic homologous recombination (NAHR). Whereas 15.6% of germline breakpoints show >20 bp of homology, this is true for only 1.1% of somatic breakpoints. This is unlikely to be an accurate estimate of absolute NAHR levels given that short-insert Illumina sequencing is biased against these events, but in a relative sense our data show that somatic NAHR is 14.2-fold less common than germline NAHR for those events that we can detect. Given that a relatively unbiased fosmid sequencing study estimated that NAHR accounts for ∼22% of germline SV (Kidd et al. 2010), our data strongly argue that recombination-based mechanisms play only a very minor role in tumor genome rearrangement. The rarity of NAHR in tumor genomes has been suggested by prior studies (Raphael et al. 2008; Hampton et al. 2009; Stephens et al. 2009; Hillmer et al. 2011). Hypotheses regarding the role of repeat-mediated homologous recombination in generating cancer genome instability (Hall and Grewal 2003; Konkel and Batzer 2010) should be reevaluated in the context of these data.
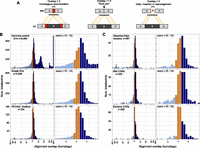
Breakpoint homology profiles. (A) When breakpoint-containing contigs are mapped to the reference genome, homology is apparent as “alignment overlap” between adjacent segments on the contig (left). “Flush” breakpoints containing no stretches of homologous DNA will have alignment overlap of approximately zero (middle). SV breakpoints harboring small insertions or small-scale rearrangements will generally have an unaligned segment, which manifests as a negative alignment overlap value (right). Occasionally, negative overlap values may also be caused by misalignment due to DNA sequencing errors or reference genome assembly errors at repeats. Overlap values are colored based on whether they are less than −1 (light blue), between −1 and 1 (orange), and >1 (dark blue). (B) Alignment overlap at germline control breakpoints (top), simple SV breakpoints (middle), and breakpoint clusters (bottom). Please note that the x-axis scale is irregular. Overlap is measured in 1-bp increments until −30 and 30, after which it is measured by tens. All breakpoints with 100 or more bases of overlap, or −100 and fewer bases, are shown at the rightmost and leftmost bars. The entire plot is shown at left and an x-axis zoom from −10 to 10 is shown at right. (C) Alignment overlap at stepwise and complex rearrangements classes following the same conventions as panel B.
We now focus on breakpoints with little or no homology. We judge variants with 2–10 bp of homology to have arisen through MMEJ or MMBIR. We judge variants with 0–1 bp of homology, or a single unaligned base (−1 bp of alignment overlap), to result from NHEJ. We consider variants with −1 to 1 bp of alignment overlap as “flush joins” given occasional alignment errors and the frequent occurrence of 1 bp of homology due to chance. It is also difficult to imagine that 1 bp of homology could function as a primer for template switching. We exclude breakpoints with 2 or more inserted bases due to the difficulty in judging whether these are due to nontemplated addition of bases during end-joining, or templated insertions during MMBIR.
Somatic breakpoints exhibit significantly less microhomology than germline breakpoints (Fig. 8B). Considering only the breakpoints with alignment overlap of −1 to 10 bp, 68% of germline breakpoints show microhomology but only 56% of somatic breakpoints do, and the distributions are significantly different (MWW; P = 2.06 × 10−39). Therefore, MMBIR and/or MMEJ are less common in tumors than in germline lineages. To our knowledge, this is the first demonstration of a difference in the utilization of microhomology-mediated mechanisms in germline versus somatic lineages.
To assess the role of microhomology-mediated processes in generating CGRs, we next compared the breakpoint homology distribution at simple SVs versus breakpoint clusters (Fig. 8B). Overall, clustered breakpoints are significantly depleted for microhomology relative to simple SV breakpoints. Whereas 57.8% of simple SV breakpoints show microhomology, only 49.2% of clustered breakpoints do, and the distribution of alignment overlap in the range of −1 to 10 bp is significantly different (MWW; P = 1.82 × 10−4). This demonstrates that MMBIR and/or MMEJ contribute significantly less to the formation of complex breakpoint clusters than to simple somatic SVs.
We next sought to address the possibility that microhomology-mediated mechanisms might be more or less common at complex versus stepwise rearrangements (Fig. 8C). Mild one-off CGRs show similar levels of breakpoint microhomology relative to simple SV breakpoints (56.4% vs. 57.8%), stepwise rearrangements show slightly less (53.4%), and chromothripsis events show dramatically less (40.3%). The only statistically significant difference is between simple SVs and chromothripsis (MWW; P = 2.01 × 10−5). These data are consistent with previous chromothripsis breakpoint sequencing experiments (Stephens et al. 2011; Kloosterman et al. 2012; Rausch et al. 2012). These data also suggest that microhomology-mediated mechanisms make a somewhat larger contribution to mild one-off CGRs than to stepwise rearrangements and chromothripsis. The reason for this is not clear. However, it is worth noting that all breakpoint clusters exhibit less microhomology than simple somatic SVs, which is precisely the opposite from what is predicted by replication-based mutational models. Thus, while this analysis does not preclude a role for MMBIR in generating CGRs, it demonstrates that MMBIR contributes less to CGR formation than it does to germline SVs and simple somatic SVs.
If a CGR is generated by template switching, then it is reasonable to expect that all of the breakpoints for that CGR would exhibit microhomology. Thus, we next asked how many variants were composed solely of breakpoints containing microhomology. Of the 134 breakpoint clusters for which one or more breakpoints were assembled, 97 (72.4%) have at least one flush breakpoint that appears to be derived from NHEJ, not MMBIR or MMEJ. This is true for 68.9% of stepwise rearrangements, 69.7% of mild one-off CGRs, and 100% of chromothripsis events. Thus, at most 27.6% of breakpoint clusters are consistent with being generated solely by microhomology-mediated mechanisms. Given that end-joining can also use microhomology through MMEJ and that MMEJ is thought to account for a nontrivial fraction of end-joining events, these data further argue that the contribution of replication-based mechanisms to CGR formation is minor.
MMBIR is thought to sometimes cause small-scale templated insertions and/or rearrangements directly at, or in the immediate vicinity of SV breakpoints, and such events should be rare for NHEJ. We thus searched for evidence of small-scale insertions and rearrangements among the assembled breakpoint-containing contigs. We find these signatures in 263 germline variants (2.2%). Given that our study is based on short-read paired-end sequencing with relatively short-insert sizes and can only detect smaller insertions/rearrangements, this measurement is roughly consistent with the ∼5% of germline SVs with breakpoint insertions previously measured by long-read sequencing (Conrad et al. 2010; Kidd et al. 2010). However, only seven clustered somatic breakpoints (0.45%) and 11 simple SV breakpoints (0.24%) show templated insertions and/or small-scale rearrangements, indicating that this signature of template switching is exceedingly rare at somatic breakpoints.
Taken together, our breakpoint profiling experiments reveal that the majority of complex rearrangements detectable in tumor genomes arise through end-joining of concurrently arising double-stranded DNA breaks, not replication-based mechanisms.
Discussion
We have performed a large-scale study of complex structural variation in 64 cancer genomes representing seven tumor types. We used a new multisample paired-end mapping algorithm to identify 6179 somatically acquired SV breakpoints, screened for complex breakpoint clusters, and profiled 4002 somatic and 6982 germline SV breakpoints at single-base resolution. To our knowledge, we have mapped a greater number of somatic breakpoints than any study to date and are the first to systematically map CGRs in a large set of tumor samples.
Our data indicate that complex rearrangements are an important aspect of cancer genome evolution. Three-fourths of the 64 cancer genomes showed at least one complex breakpoint cluster, and one-quarter of all breakpoints were found in clusters. Based on copy number state profiling, 63% of clusters are consistent with originating through a one-off mutational event, and these comprise 13.6% of all somatic breakpoints discovered in this study. Thus, our data argue that although the absolute number of complex mutational events is relatively low, representing just 1.8% of all structural mutations, these events have a large and diverse genomic impact.
The availability of 64 diverse cancer genomes generated by a single sequencing platform allowed us to assess the frequency of chromothripsis among tumor types. Previous studies have focused on one tumor type or have relied on microarrays, which are poorly suited to detecting CGRs. We identified chromothripsis events in an unbiased, automated fashion and found a significantly higher incidence in GBM (38.9%) relative to the other tumor types (8.7%). This definitively shows that chromothripsis is a variable phenotype among tumor types. At present, it is unclear whether variable prevalence is due to differences in the frequency of specific trans-acting mutations, variable exposure to chromothripsis-causing mutagens, differences in the selective pressures faced by different cancers, and/or other unknown factors. Additional work will be required to resolve this important question.
Finally, our results help resolve the key mechanistic question of how CGRs arise. Unlike previous studies, we have assessed the entire spectrum of complex rearrangements, from mild CGRs to staggeringly complex chromothripsis variants, and we have characterized an extremely large number of breakpoint sequences. Our data provide strong evidence that complex tumor genome rearrangements are formed predominantly through end-joining, not microhomology-mediated break-induced replication (MMBIR). We therefore propose that most CGRs arise when multiple double-strand DNA breaks exist at the same time, in the same cell, and that the fundamental difference between chromothripsis events and milder forms of complex rearrangement is the severity of the original DNA damage event. The observed prevalence of complex rearrangements further implies that the simultaneous generation of multiple spatially clustered DNA breakages is an alarmingly common occurrence and begs the question of what environmental mutagen or cellular process is responsible for this damage.
Methods
Variant detection
TCGA data sets generated by Illumina paired-end sequencing were downloaded from dbGAP as BAM files. Discordant read pairs were extracted separately for each read group and re-aligned to the reference genome (NCBI Build 37) with Novoalign using sensitive settings (-k 14 -s 1). Repetitive alignments were resolved using the “random” mode (-R). For each data set, discordant mappings for each read group were converted to BEDPE format (Quinlan and Hall 2010) and combined into a single file, and duplicates were removed with dedupDiscordantsMultiPass (http://code.google.com/p/hydra-sv/) allowing inexact coordinate matching (-s 3). Read groups were then classified into their initial genomic libraries using insert size statistics.
Breakpoints were detected with HYDRA-MULTI, a new multisample version of HYDRA (Quinlan et al. 2010). In essence, all discordant mappings from all data sets are pooled prior to breakpoint calling, and presence/absence genotypes are calculated based on the number of read pairs from each data set that form the call (Quinlan et al. 2011). A configuration file was prepared detailing the insert size distribution of each of the 377 total sequencing libraries from the 129 data sets.
A total of 4,686,652 breakpoints were predicted, all but 1,636,145 of which were due to a previously unreported library preparation artifact that produces a profuse number of false small (<10 kb) inversion calls. We therefore removed all inversion calls smaller than 10 kb. Paired-end mapping is prone to false positives due to read mapping artifacts and reference genome assembly errors, and thus we also required breakpoint calls to fulfill the following criteria: (1) at least three read pairs support the call; (2) the reads have a mean mapping quality >30; (3) the reads have a mean number of mappings <1.5; (4) the variant call is at least 100 bp in size; and (5) neither end of the call overlaps simple or satellite repeats by >50% (bedtools pairtobed -type either -f 0.5), as defined by a union of the UCSC “simpleRepeat” track and the simple and satellite repeat annotations present in the “RepeatMasker” tracks. These filtering steps resulted in 34,621 high-confidence calls, 6179 of which were judged to be somatic by their presence in one tumor sample and none of the remaining 128 samples.
To identify known deletions within our data set, we compared our deletion calls to validated deletions from the 1000 Genomes Project (Mills et al. 2011). To compare calls, we used bedtools intersect requiring 50% reciprocal overlap (-r -f 50).
Assembly and validation
We modified the sga walk function from the String Graph Assembler (SGA) (Simpson and Durbin 2012) to report all walks from all connected components of the string graph. For each breakpoint call, we extracted all read pairs for which either read mapped within 500 bp and ran the following commands: sga preprocess (default), sga index (-no -reverse), sga correct (-k 13 -x 2 -d 128), sga index on the error corrected reads (default), sga rmdup (default), sga overlap (-m 15), sga assemble (-m 15 -d 0 -g 0 -b 0 -l 100), and our modified version of the sga walk program (-d 10000–component-walks). Contigs were aligned to the reference using BWA-SW (Li and Durbin 2010). Split-mappings with ≥25 bp of nonoverlap with an adjacent mapping on the contig were converted to BEDPE format and compared with HYDRA-MULTI calls using bedtools pairtopair (-type both). We judged a call validated if the breakpoints predicted by split-mapping overlapped with the 200-bp breakpoint intervals predicted by HYDRA-MULTI and were the same variant class.
Identification of breakpoint clusters
Breakpoints from a single sample within 100 kb of each other were merged using bedtools cluster (-d 100000), and “breakpoint clusters” were formed by chaining together genomic regions sharing one or more breakpoint calls. The result is a set of breakpoint clusters in which each breakpoint is present in one cluster and all genomic regions within a cluster are linked by a series of breakpoint calls. We retained clusters composed of three or more breakpoints and merged clusters found within 1 Mb of each other. For all simulations, we report the mean of 100 replicates. For the Monte Carlo simulation, somatic breakpoint coordinates were randomized using bedtools shuffle, excluding assembly gaps. To emulate the filtering of real breakpoint calls, shuffled breakpoints were only placed within uniquely mappable regions of the genome, as defined by the UCSC wgEncodeCrgMapabilityAlign100mer track, and were not allowed to overlap with simple or satellite repeats defined by the UCSC RepeatMasker track. For the other simulations, sets of various SV callsets were randomly sampled to match the number of somatic SVs identified in each tumor. To measure enrichment of genome annotations at breakpoint clusters, we calculated the observed overlap divided by the median of a Monte Carlo simulation conducted with pybedtools (100 trials) (Dale et al. 2011).
Read-depth analysis
We used bedtools coverage to measure read depth in genomic windows containing 5 kb of uniquely mappable sequence. We corrected for GC bias using a normalization procedure that expresses read depth as a Z-score calculated from windows with similar GC content (Quinlan et al. 2010). To estimate copy number, we divided read depth by the median read depth of all other windows with a similar GC content, and multiplied by 2. To detect CNAs, we performed circular binary segmentation (Olshen et al. 2004) using the R DNAcopy package (undo.splits=”sdundo” and undo.SD=2). We defined CNA change-points as the interval (±5 kb) between adjacent CNA segments whose median Z-score differed from each other by >1 median absolute deviation. We defined somatic change-points as those found in a single tumor sample but not in the 65 normal samples, requiring 100% reciprocal overlap between change-points (bedtools intersect -r -f 1) and the same direction of copy number change.
To compare change-points and breakpoints, we defined overlap as being within 10 kb. To compare CNA change-points and breakpoint clusters, we defined overlap as being within 50 kb. To measure enrichment, we compared the observed overlap with the mean found in Monte Carlo simulations in which breakpoints and breakpoint clusters, respectively, were randomly shuffled 100 times.
To estimate copy number states at breakpoint clusters, we extracted CNA change-points within 100 kb. We used this generous definition to compensate for imprecise change-point detection and false-negative breakpoint calls, thus helping to ensure that the number of CNA states was not underestimated. We then used a custom CNA state determination algorithm that operates on a sorted list of predicted copy numbers taken from change-points. The algorithm merges change-point values into a group if the smaller value is at least 80% of the larger value and then recalculates the copy number by taking the mean of the values in the group. The only exception is that the two copy number values for a given change-point cannot be placed into the same group. In this case, a new group is initiated, and the process is repeated for the remaining values. We chose this greedy algorithm after testing more conventional methods including k-means clustering, hierarchical clustering, and kernel density estimation. These methods routinely underestimated the number of copy number states at a nontrivial fraction of breakpoint clusters, leading to misclassification of stepwise rearrangements as CGRs.
Monte Carlo simulation of progressive rearrangement
To assess the likelihood that an extreme CGR was due to one-off rather than stepwise mutation, we performed a simulation based on the method of Stephens et al. (2011). For each observed CGR, we performed a Monte Carlo simulation in which the observed SV breakpoint calls were applied in random order to a progressively mutated synthetic chromosome. We estimated the probability that the observed CGR is caused by stepwise rearrangement (the null hypothesis) by dividing the number of simulation runs that produced the same or fewer copy number states as observed in the real data by the total number of successful simulation runs.
To perform simulations, we used a modified version of SVsim (G Faust, unpubl.), a structural variation simulator. We simulate rearrangements on multiple chromosomes, but we do not allow rearrangements between chromosomes. We use a diploid genome for our simulations to more accurately mirror natural conditions and to help mitigate the loss of genomic regions via deletions. This is more conservative than a haploid simulation in that it generally results in fewer CNA states. During the simulation, we take into account the orientation of mutated chromosomal segments when determining the relationship between read-pair orientation and event type. To select breakpoint locations within multicopy regions generated by a prior duplication, we randomly select one of the breakpoint loci and then select the second locus that is closest to it on the mutated chromosome. If a breakpoint cannot be applied due to a prior deletion, we attempt to apply the rearrangement to the homologous chromosome; if it cannot be applied to the homolog, we abort the simulation run and try again. At the end of each successful run, we count the number of distinct copy number states across the entire mutated genome. As our ability to observe copy number states in actual data is restricted to the resolution of our read-depth analysis, we only count states in our simulations that appear in regions exceeding 10 kb in length. We continue this process until 1000 successful simulation runs have completed for each CGR.
Identification of templated insertion events
To identify templated insertion events and small-scale rearrangements at SV breakpoints, we examined contigs that validated a HYDRA-MULTI call and contained ≥20 bp of unaligned sequence directly at the breakpoint, as determined by BWA-SW. We aligned these contigs to the reference with YAHA (Faust and Hall 2012) (k-mer size 15, -M 15 -P 0.8 -H 5000). We visualized alignments with a modified version of PARASIGHT (J Bailey and E Eichler, unpubl.; http://eichlerlab.gs.washington.edu/jeff/parasight) and scrutinized breakpoints for insertions derived from elsewhere in the genome, as well as for small-scale rearrangements directly at the breakpoint.
Estimating intra-tumor breakpoint allele frequency (BAF)
For each breakpoint-containing contig, we extracted 200 bp of sequence flanking the breakpoint and aligned the raw reads from each data set using BWA (default). To consider an alignment as positively genotyping the variant allele, we required that it spanned the breakpoint with at least 20 bp on both sides. To genotype the reference allele, we extracted the 200 bp flanking each of the two breakpoint positions in the reference genome and performed alignment as above. BAF is given by the number of reads aligning to the variant junction divided by the mean number of reads aligning to the two reference junctions. To consider a BAF measurement as sufficiently precise for subsequent comparative analyses, we required that at least three reads identified the variant allele.
Statistical analyses
Statistics were performed in MATLAB. When multiple Mann-Whitney-Wilcoxon (MWW) tests were used to compare the breakpoint allele frequency or homology for different variant classes, we state the Bonferroni-corrected P-value.
Acknowledgments
We thank J. Simpson for advice regarding modifications to the SGA assembler and the TCGA Network for producing the data used in this study. This work was supported by a DoD Breast Cancer Post-doctoral Fellowship to A.M.; an NIH/NHGRI grant (1R01HG006693-01) to A.R.Q.; and an NIH New Innovator Award (DP2OD006493-01), a March of Dimes Basil O'Connor Research Award, and a Burroughs Wellcome Fund Career Award to I.M.H.
Footnotes
-
↵5 Corresponding authors
E-mail irahall{at}virginia.edu
E-mail arq5x{at}virginia.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.143677.112.
- Received May 25, 2012.
- Accepted February 12, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








