
DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation
- Anne Y. Lai1,
- Deepak Mav2,
- Ruchir Shah2,
- Sara A. Grimm3,
- Dhiral Phadke2,
- Katerina Hatzi4,
- Ari Melnick4,
- Cissy Geigerman5,
- Steve E. Sobol6,
- David L. Jaye5 and
- Paul A. Wade1,7
- 1Laboratory of Molecular Carcinogenesis, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina 27709, USA;
- 2SRA International, Inc., Durham, North Carolina 27709, USA;
- 3Integrative Bioinformatics, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina 27709, USA;
- 4Weill Cornell Medical College, New York, New York 10065, USA;
- 5Department of Pathology and Laboratory Medicine,
- 6Department of Otolaryngology–Head and Neck Surgery, Emory University School of Medicine, Atlanta, Georgia 30322, USA
Abstract
Memory is a hallmark of adaptive immunity, wherein lymphocytes mount a superior response to a previously encountered antigen. It has been speculated that epigenetic alterations in memory lymphocytes contribute to their functional distinction from their naive counterparts. However, the nature and extent of epigenetic alterations in memory compartments remain poorly characterized. Here we profile the DNA methylome and the transcriptome of B-lymphocyte subsets representing stages of the humoral immune response before and after antigen exposure in vivo from multiple humans. A significant percentage of activation-induced losses of DNA methylation mapped to transcription factor binding sites. An additional class of demethylated loci mapped to Alu elements across the genome and accompanied repression of DNA methyltransferase 3A. The activation-dependent DNA methylation changes were largely retained in the progeny of activated B cells, generating a similar epigenetic signature in downstream memory B cells and plasma cells with distinct transcriptional programs. These findings provide insights into the methylation dynamics of the genome during cellular differentiation in an immune response.
Epigenetic modifications play important roles in regulating cellular differentiation events. One such epigenetic modification, DNA methylation, occurs on cytosine residues primarily at CpG dinucleotides in mammals. The role of DNA methylation in regulating cellular differentiation from pluripotent and multipotent progenitors has been demonstrated through functional analysis of animals deficient in DNA methyltransferases (DNMTs) (Li et al. 1992; Okano et al. 1999; Tadokoro et al. 2007; Broske et al. 2009; Wu et al. 2010), as well as from recent genome-wide studies comparing the DNA methylome of various differentiated cell types and their precursors (Meissner et al. 2008; Lister et al. 2009; Ji et al. 2010; Hodges et al. 2011; Bock et al. 2012). In the context of the immune system, mutations in the DNMT3B gene are causal for the development of ICF syndrome (immunodeficiency, centromere instability, and facial anomalies syndrome), a rare autosomal recessive immune disorder (Hansen et al. 1999; Xu et al. 1999). Despite having a normal number of mature B cells, ICF patients lack memory B cells as well as plasma cells (PCs) (Blanco-Betancourt et al. 2004), suggesting the involvement of DNMT3B and possibly of DNA methylation in regulating late stages of lymphocyte maturation.
Upon activation by antigenic stimulation in a T-cell–dependent B-cell immune response, naive B cells enter the germinal-center (GC) reaction in secondary lymphoid organs. Within GCs, B cells activated by antigenic stimuli clonally expand and their immunoglobulin (Ig) gene loci are subjected to somatic hypermutation and class-switch recombination (Victora and Nussenzweig 2012). These genetic alterations are critical for the maturation of GC B cells to post-GC cell types that subsequently produce high-affinity antibodies against foreign pathogens. Upon exiting the GC, B cells either differentiate into antibody-producing long-lived PCs or alternatively become memory B cells that provide long-term immunity against the same pathogen (Shapiro-Shelef and Calame 2005). During a secondary immune challenge, memory B cells more rapidly undergo a proliferative burst, and then differentiate into PCs in a facilitated manner compared to naive B cells (McHeyzer-Williams and McHeyzer-Williams 2005; Lanzavecchia and Sallusto 2009). Compared to naive B cells, the memory counterparts express B-cell receptors with higher affinity to the same antigen (Pascual et al. 1994), constitutively express costimulatory molecules on their cell surface (Liu et al. 1995), and have lower expression of transcription factors (TFs) important for maintaining cellular quiescence (Good and Tangye 2007). These unique features decrease the threshold of activation in memory B cells and allow them to quickly enter the cell cycle upon restimulation. Aside from these key differences, naive and memory B cells possess highly similar gene expression programs (Klein et al. 2003), and it remains unclear how memory B cells can more efficiently reprogram their transcriptional profiles to specify a PC fate. It has been speculated that epigenetic alterations in naive and memory lymphocytes contribute to their functional outcomes (Messi et al. 2003; Kersh et al. 2006; Cuddapah et al. 2010). Nonetheless, the degree of epigenetic differences in these two cell types remains undefined. It is also unclear whether DNA methylation plays a role in specifying an effector vs. a memory cell fate in lymphocytes during a humoral immune response.
The global methylation landscape of the total B-cell fraction in peripheral blood was previously characterized, revealing distribution of this epigenetic mark at different genomic features (Rauch et al. 2009). To further understand the dynamics of DNA methylation changes during a B-cell immune response, naive, GC, memory, and PC populations were purified ex vivo from inflamed tonsils of eight individual humans for global DNA methylation analysis. We observed widespread DNA methylation changes between naive and GC B cells. In contrast, fewer changes in DNA methylation occur upon resolution of activated, GC B cells into either memory or PC alternative cell fates. These results demonstrate that memory B cells and PCs share a highly similar methylome, despite having very distinct transcriptional programs and that naive and memory B cells, which have highly similar transcriptomes, have vastly different methylomes. In agreement with genome-scale studies in other cell types (Stadler et al. 2011; Bock et al. 2012; Thurman et al. 2012), differentially methylated regions (DMRs) in GC B cells were enriched for TF binding sites or regulatory elements. A second class of loss-of-methylation events localized to Alu elements, consistent with the preferential targeting of DNMT3 family de novo methyltransferases to repetitive elements in the genome (Jeong et al. 2009). The methylation dynamics at Alu elements suggests a regulatory role of the repetitive fraction of the genome in the GC transcriptional program and/or its associated chromatin architecture, and a potential contribution to genomic instability in GC B cells (Ranuncolo et al. 2007). We propose that global reprogramming of the epigenome during the GC reaction represents an event that permits an adult somatic cell to differentiate into multiple, diverse cell types. In addition, the epigenetic signature of memory B cells may contribute to their superior ability, compared to that of naive B cells, to rapidly differentiate into PCs upon antigen rechallenge during a secondary immune response.
Results
Widespread alteration of the DNA methylation landscape in activated B cells is conserved in post-GC cell types
Naive, GC, memory B cells, and PCs were purified from inflamed tonsils by FACS (Fig. 1A,B). The four cell types can be distinguished by IgD and CD38 cell surface expression within CD20+ B cells (Pascual et al. 1994; Klein et al. 2003). In monitoring expression of key molecules by quantitative RT-PCR to validate cell purity, we observed significant changes in expression of all three mammalian DNMTs (DNMT1, DNMT3A, and DNMT3B) (Jones and Liang 2009) in B-cell subsets during the course of an immune response (Fig. 1C). While the expression level of DNMT3A dramatically decreased upon activation by antigen (naive to GC B-cell transition), DNMT1 and DNMT3B were up-regulated at this stage, consistent with the role of DNMT1 in replication-dependent maintenance methylation (Leonhardt et al. 1992) and with the role of DNMT3B in late-stage B-cell differentiation (Blanco-Betancourt et al. 2004). Post-GC memory B cells and PCs reverted the expression pattern of DNMTs to that of their naive precursors (Fig. 1C). Immunohistochemical staining of tonsil sections confirmed robust expression of DNMT1 and minimal expression of DNMT3A in GC B cells (Fig. 1D). In contrast, regions surrounding the GC comprised primarily of naive B cells have the opposing expression pattern of the corresponding DNMT. Up-regulation of DNMT3B in GC B cells at the protein level was also confirmed by immunofluorescence (Fig. 1E). Expression changes in all three forms of DNMTs upon immune activation pointed to possible reprogramming of the DNA methylation landscape in GC B cells.
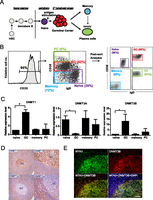
Expression pattern of DNMTs in tonsillar B-cell subsets. (A) Cartoon illustrating the stages of B-cell development and T-dependent B-cell immune response. B cells are differentiated from hematopoietic stem cells (HSCs) in the bone marrow and released into the periphery upon maturation. Naive B cells encounter antigen-primed T cells in secondary lymphoid organs and enter the germinal center (GC) reaction followed by differentiation into memory B cells or antibody-secreting plasma cells (PCs). (B) Purification of B-cell subsets. CD20, IgD, and CD38 cell surface expression were used to distinguish the cell populations: Naive B cells (CD20+IgD+CD38lo), GC B cells (CD20+IgD−CD38+), memory B cells (CD20+IgD-CD38−), and PCs (CD20lo/+IgD−CD38hi). The percentage of each subset within tonsillar B cells is shown in the FACS plot in the middle panel. Naive, GC, memory, and PC populations were purified from eight individuals for gene expression and DNA methylation profiling on microarrays. The purity of each cell population (>90%) was confirmed by flow cytometry analysis of post-sorted cells (right panel). (C) Transcript abundance of DNMTs in B-cell subsets by quantitative RT-PCR. DNMT expression levels are normalized to actin expression level in each cell type. The expression level in one biological replicate of naive B cells is arbitrarily set to one. Error bars denote standard deviation of three biological replicates. (D) Expression patterns of DNMT1 and DNMT3A in tonsils. (Upper left, upper right) Immunohistochemical staining of DNMT1 at 200× and 400× magnifications, respectively. (Lower left, lower right) Immunohistochemical staining of DNMT3A at 200× and 400× magnifications, respectively. (E) Up-regulation of DNMT3B in GC B cells. Tonsil sections were costained with anti-DNMT3B (red) and anti-MTA3 (green). Nuclei were labeled with DAPI (blue). DNMT3B expression colocalized with MTA3, which is a marker for GC B cells (Fujita et al. 2004). Scale bar, 50 μm.
To examine the dynamics of DNA methylation during the immune response, global DNA methylation profiling was performed on the four tonsillar B-cell subsets from eight individuals using the methylated CpG island recovery assay (MIRA) (Rauch et al. 2006) and analyzed on microarrays (MIRA-chip). While the number and genomic coverage of autosomal regions enriched in DNA methylation peaks varied by cell type (Supplemental Table S1) and also by subject (data not shown), DNA methylation dynamics relevant to immune activation and the subsequent cell differentiation programs were presumed to be consistent across all individuals. DMRs were identified for each pair of cell types across all subjects to assess the DNA methylation changes during the immune response (Supplemental Table S2). There were a greater number of DMRs associated with immune activation, at the transition from naive to GC B-cell stage (Fig. 2A; Supplemental Table S2), than with any other developmental transition. Notably, immune activation-induced DMRs were dominated by loss-of-methylation events (Fig. 2A), in agreement with a previous study profiling the status of 50,000 CpGs during activation (Shaknovich et al. 2011). Substantially fewer DMRs were identified between GC B cells and memory B cells or PCs (Fig. 2A; Supplemental Table S2). DNA methylation levels at activation-induced DMRs were highly similar when comparing GC, memory B cells, and PCs (Fig. 2B, left panel). Principle component analysis (PCA) also indicated that the methylation signature of GC and post-GC cell types cluster closely together, clearly distinct from naive B cells (Fig. 2B, right panel). These data indicate that the majority of activation-induced DNA methylation changes in the GC are inherited by both memory B cells and PCs. Loss-of-methylation events in GC B cells were equally distributed near promoter-proximal regions and promoter-distal regions (Fig. 2C,D). In contrast, gain-of-methylation events were overrepresented in promoter-proximal regions (Fig. 2C,D). The genomic distribution between the two types of DMRs is significantly different by the χ2 test, P < 0.0001, indicating the possibility of distinct regulatory roles of gain- and loss-of-methylation events.
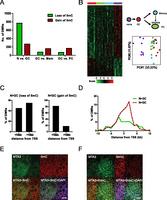
Widespread alteration of DNA methylation in B-cell subsets. (A) Assessment of global DNA methylation changes during immune activation (naive [N] vs. GC) and differentiation of GC B cells into memory or PCs (GC vs. memory [Mem] and GC vs. PC) by MIRA-chip analysis. The bar graph displays the number of DMRs with increased methylation (gain of 5mC) in red or decreased methylation (loss of 5mC) in green in each stage transition. (B) The methylation score is computed for each DMR between naive and GC B cells in all MIRA-chip samples and displayed in a heat map (left panel). Each row represents a DMR between naive and GC. Each column represents an individual MIRA-chip sample. All eight samples from each cell type clustered together in the heat map. Cell types are represented by different colors, as indicated in diagram beside the heat map. PCA analysis of methylation score of DMRs for all cell types is shown in the right panel. Each circle represents an individual biological sample. x- and y-axes represent first and second principal components, respectively. The degree of variance in each principal component is indicated in parentheses. (C) Frequency and genomic distribution of DMRs between naive and GC B cells. The bar graphs display the number of DMRs that are <10 kb or >10 kb from the nearest annotated TSS. The left panel represents DMRs with loss of methylation between naive and GC B cells (N > GC), while the right panel represents DMRs with gain of methylation between naive and GC B cells (N < GC). (D) Distribution of DMRs relative to the nearest TSS. The frequency of both types of DMRs, loss-of-methylation DMRs (N > GC), and gain-of-methylation DMRs (N < GC), in 1-kb increments from the TSS is shown in the graph. (E) Distribution of methylcytosine (5mC) in tonsils. A tonsil section was double stained for MTA3 to demarcate GC B lymphocytes (green) and 5mC (red). Nuclei were labeled with DAPI (blue). There was no detectable difference in the global level of 5mC in MTA3+ GC B cells and in non-GC B cells. Scale bar, 50 μm. (F) Distribution of hydroxy-methylcytosine (5hmC) in tonsils. A tonsil section was double stained for MTA3 to demarcate GC B lymphocytes (green) and 5hmC (red). Nuclei were labeled with DAPI (blue). The global level of 5hmC decreased in in MTA3+ GC B cells compared to non-GC B cells. Scale bar, 50 μm.
Widespread loss-of-methylation could be a result of passive or active demethylation (Wu and Zhang 2010). Two major active demethylation pathways have been established in mammals: (1) activation-induced cytosine deaminase (AICDA)/APOBEC-dependent deamination of 5-methyl-cytosine (5mC) followed by base excision repair; and (2) oxidative demethylation by TET proteins. Although AICDA is highly expressed in the GC, recent genome-wide analysis of AICDA occupancy indicates a strong preference for promoter-proximal regions (Yamane et al. 2011). However, we did not observe a biased distribution of loss-of-methylation DMRs near TSSs in GC B cells (Fig. 2C,D). Further, AICDA/APOBEC enzymes have substantially reduced activity on 5mC relative to unmodified cytosine (Nabel et al. 2012). Therefore, AICDA may not be the mediator of the alterations observed here.
To explore the TET pathway in this system, we examined the global level of 5hmC, the product of TET proteins, in naive and GC B cells by immunofluorescence. While the level of 5mC remained unchanged in the two B-cell subsets (Fig. 2E), we observed a decrease in 5hmC in GC B cells (Fig. 2F), arguing against the role of this demethylation pathway in the GC. The most likely explanation for the site-specific DNA methylation changes observed in GCs is passive demethylation rather than an active process. We envision two possible explanations that are not mutually exclusive: (1) Declines in DNMT3A levels coupled with a massive burst of proliferation lead to a failure to maintain DNA methylation levels; and (2) global alteration in nuclear architecture and nucleosome positioning results in a change in substrate quality for DNMTs.
B-lymphocyte activation establishes DNA methylation reprogramming that poises memory B cells for recall responses
To gain insight into the relationship between DNA methylation and gene expression, we performed global transcriptional profiling in the purified cell populations used for DNA methylation analysis. Selected genes diagnostic of the developmental transitions occurring during the immune response were manually validated by RT-PCR and inspected from the microarray data (Supplemental Fig. S1A,B). Unsupervised hierarchical clustering and principal component analysis (PCA) of the gene expression profiles indicated that naive and memory B cells share a highly similar transcriptional program, but are distinct from GC as well as PC cell types (Fig. 3, far left panels). More than 1200 genes were differentially expressed between each pair of cell types examined, the exception being the comparison between naive and memory cells where only 158 genes were differentially expressed (Supplemental Fig. S1C).
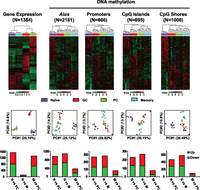
B-lymphocyte activation establishes DNA methylation reprogramming that poises memory B cells for recall responses. (Top) Heat maps displaying hierarchical clustering of genes differentially expressed (ANOVA P < 0.01 and fold change > 3) and genomic features with differential methylation scores (ANOVA P < 0.01). The total number of genes or genomic features is indicated. (Middle) PCA analysis of the corresponding differentially expressed genes or differentially methylated genomic features. Each circle represents an individual MIRA-chip sample; cell types are represented by different colors. x- and y-axes represent first and second principal components, respectively. The degree of variance in each principal component is indicated in parentheses. (Bottom) Analysis of genomic features by average score in naive B cells (N) vs. PC; naive B cells (N) vs. memory B cells (M); and memory B cells (M) vs. PC. The bar graph displays the number of genomic features with increased methylation (up) in red and decreased methylation (down) in green.
The extent of DNA methylation difference between the four B-cell subsets was further investigated using a mixed-effect model to analyze the differences in average methylation score at various defined genomic features (Weber et al. 2007). Strikingly, the close similarity of memory and naive cell populations in terms of transcriptional profile contrasts sharply with the pattern of DNA methylation at all genomic features examined. Although there were differentially methylated genomic features between memory B cells and PCs, these two cell types clustered closely at most features (Alus, CpG islands, and CpG island shores) (Fig. 3, top panel), suggesting a high degree of similarity between the two methylomes. The first principal component of PCA resolved GC B, memory B, and PC from naive B cells for all four genomic features, while a much smaller degree of variance was detected between GC, memory B, and PCs (Fig. 3, middle panel). The genomic feature analysis was consistent with the DMR analysis (Fig. 2B; Supplemental Table S2), suggesting that DNA methylation is globally altered upon activation in naive B cells, while the dynamics are more subtle as cells partition between memory B-cell vs. PC compartments as their post-GC fate. In agreement with hierarchical clustering and PCA, there are significantly fewer differentially methylated genomic features between memory B cells and PCs, compared to naive vs. PCs or naive vs. memory B cells (Fig. 3, bottom panel; Supplemental Tables S2, S3). These data demonstrate that antigen-driven activation in B cells induces global DNA methylation reprogramming, resulting in a common epigenetic signature in memory B cells and PCs, despite the adoption of very different transcriptional programs in post-GC cell types.
To determine whether the DNA methylation profiles described here may contribute to the functional superiority of memory cells in differentiation into PCs, we investigated whether genomic regions differentially methylated between naive and memory B cells are associated with genes or regulatory elements with known functions in immune responses. DMRs between naive and memory B cells that were associated with genes (within 10 kb of TSS) were divided into two categories: naive > memory or naive < memory in methylation signal. Both categories were enriched for genes with functions associated with immune response and hematological functions, gene expression, cell growth and proliferation, and cellular functions integral to lymphocyte activation (Supplemental Table S4). Genomic feature analysis at promoters (−1 to +1 kb from TSS) and CpG island shores (located within 10 kb of gene promoters) revealed highly similar enrichment in functions relevant to immune response (data not shown). Exemplar genes having immune-related functions that differ in methylation pattern (Supplemental Fig. S2A) include critical regulators of B-cell development (PAX5), of immune responses (RELA), and of cell death/survival (BCL2L1 and RUNX3). Importantly, the expression level of these genes, while similar in naive and memory B cells, changes significantly upon differentiation into PCs (Supplemental Fig. S2B). The association of DNA methylation changes with elements associated with immune activation and the immune response supports the concept that the memory B-cell epigenome is poised to facilitate a more rapid and robust activation response than their naive counterparts.
Enrichment of regulatory elements and TF binding sites at activation-induced DMRs
To further investigate the function of DNA methylation changes associated with immune activation, we first determined whether activation-induced DMRs were enriched for regulatory elements. The degree of overlap between DMRs and DNase I hypersensitive sites (DHSs) from ENCODE DNase I-seq data sets (Thurman et al. 2012) was queried. Both loss-of-methylation and gain-of-methylation DMRs were highly enriched for DNase I clusters, which consist of combined DHS identified across 125 diverse cell and tissue types (Fig. 4A; Thurman et al. 2012). The majority of the DHS overlapping DMRs were present in primary B cells (Fig. 4A), consistent with a relationship between DNA methylation and regulatory factor occupancy patterns (Stadler et al. 2011; Bock et al. 2012; Thurman et al. 2012). We further searched for the presence of DNA binding motifs of several TFs important for B-cell development within the DMRs (Table 1). In line with the DNase I HS analysis, we identified several TFs, including MYC and BCL11A, with motifs that were significantly enriched within loss-of-methylation DMRs in GC B cells (Fig. 4B; Table 1). Analysis of MYC by ChIP-PCR (Supplemental Fig. S3A) and of MYC and BCL11A ChIP-seq data from ENCODE (The ENCODE Project Consortium 2011) in a B-lymphoblastoid cell line indicates that the DMRs frequently overlap with the binding sites of these TFs (Fig. 4B; Supplemental Fig. S3A). Further strengthening the connection between DMRs and regulatory DNA, we observed increased levels of histone 3 lysine 27 acetylation (H3K27ac), consistent with these regions being active enhancers (Creyghton et al. 2010), in GC B cells at several loss-of-methylation DMRs (Supplemental Fig. S3B).
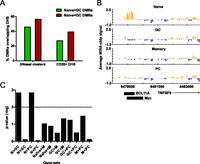
Activation-induced DMRs reveal cis-regulatory elements in GC B cells. (A) Overlap of DMRs with DNase I hypersensitive sites (DHSs). The percentage of loss-of-methylation (N > GC) or gain-of-methylation (N < GC) DMRs intersecting DNase I clusters from any cell type or DHS from primary CD20+ B cells is shown. (B) Example of a DMR overlapping a DNase I HS as well as MYC and BCL11A binding sites in a lymphoblastoid cell line. The bar graphs display the average MIRA-chip signals from eight biological replicates in naive, GC, memory, and PC cells at the genomic region on chromosome 19, as indicated on the x-axis. The location of an annotated gene (TNFSF9) is shown below the bar graphs relative to the genomic coordinates on chromosome 19, and the orientation of the arrow indicates the direction of transcription. Black rectangles below the plots indicate the location of MYC and BCL11A binding sites determined by ChIP-seq data from the GM12878 lymphoblastoid cell line (performed by Iyer's group at University of Texas-Austin and Myer's group at HudsonAlpha, respectively) as determined by the ENCODE project (The ENCODE Project Consortium 2011). (C) Analysis of the association between DNA methylation and gene expression changes by GSEA. The –log of enrichment P-value for each gene set (differentially expressed genes in each pair of cell types) is shown. Gene sets with –log of P-value above the dotted line (>2 or P < 0.01) are considered to be statistically significant.
Transcription factor motif analysis of DMRs between naive and GC B cells
The relationship between DNA methylation dynamics and gene expression was further examined. Gene set enrichment analysis (GSEA) (Subramanian et al. 2005) revealed that genes with down-regulated transcription during the naive to GC or the naive to PC differentiation events were significantly enriched for those that have overall gained methylation in GC B cells (Figs. 4C; Supplemental Table S5). PAX5, a TF critical for B-lineage commitment whose down-regulation is required for the activation of the PC transcriptional program (Delogu et al. 2006), exemplifies genes gaining methylation on activation (Supplemental Fig. S2B). These results are consistent with the observation that gain-of-methylation DMRs are preferentially enriched near transcription units and within genic regions (Fig. 2D). We did not observe a significant association between loss of DNA methylation on activation and gene expression, perhaps reflecting the likelihood that many of these methylation losses occur at distal regulatory DNA that is difficult to assign to a particular gene. In any case, these analyses indicate that DNA methylation dynamics in B-cell subsets representing various stages of the immune response provide insights into the regulatory elements that govern the GC reaction and transcriptional program.
Widespread loss of DNA methylation at Alu elements on activation
About half of activation-induced DMRs overlapped with DHS (Fig. 4A), the mechanistic underpinning of the remaining DMRs was unclear. When mapping DMRs to various genomic features, a large proportion of DMRs between naive and GC B cells were adjacent to Alu elements (Fig. 5A; Supplemental Table S2). This observation is contrary to the widely held view that methylation at repetitive elements is constitutive in normal somatic cell types (Hellmann-Blumberg et al. 1993). Loss-of-methylation DMRs are especially predominant over gain-of-methylation events near this genomic feature (Fig. 5A).
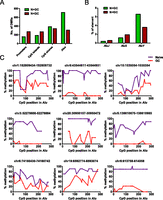
Widespread loss of DNA methylation at Alu elements in GC B cells. (A) Distribution of DMRs between naive and GC B cells at various genomic features (for definition of each genomic feature, see Methods). The bar graph illustrates the number of loss-of-methylation DMRs (naive > GC) in green and gain-of-methylation DMRs (naive < GC) in red that overlap with the indicated genomic feature. (B) Percentage of elements within each Alu subfamily (J, S, and Y) with loss (N > GC) or gain (N < GC) of methylation in GC compared to naive B cells by MIRA-seq analysis. (C) Validation of differentially methylated Alu elements in naive and GC B-cell populations by genomic bisulfite sequencing. The methylation status of CpGs across nine Alu elements was analyzed. After bisulfite treatment and PCR amplification of the Alus, at least eight individual clones of the PCR product were analyzed. The percentage of methylated cytosine (y-axis) was calculated at individual CpG positions relative to the 5′ end of each Alu element (x-axis). The genomic location (hg19) of the Alu elements analyzed are listed above each graph.
To explore the prevalence of loss-of-methylation at Alu elements in GC B cells, we analyzed the average methylation score at each Alu element covered in our experiments (Fig. 5A; Supplemental Fig. S4). Since the microarrays lack coverage within repetitive elements, we used probes adjacent to repeats (within 250 bp) as a surrogate for their methylation status in subsequent analysis as these closely correlated with the methylation levels within candidate Alu elements assessed by genomic bisulfite sequencing (Supplemental Fig. S4). In agreement with the DMR analysis, differentially methylated Alus lost methylation in GC B cells and remain hypomethylated in post-GC cell types (Fig. 3). PCA analysis of differentially methylated Alus again indicated dissimilarity between naive B cells and all other cell types arising after immune activation (Fig. 3).
We further investigated the dynamics of Alu methylation in naive and GC B cells at the base pair level, by performing next generation sequencing of bisulfite-converted MIRA-enriched DNA fragments from naive and GC B cells (MIRA-seq) (Serre et al. 2010). The AluY subfamily of elements had the highest sequencing coverage (Supplemental Table S6), reflective of the high CpG content within this Alu family (Supplemental Fig. S5A). Since the majority of MIRA-enriched CpG dinucleotides were methylated (Supplemental Table S7), we identified differentially methylated Alus solely based on sequencing depth. Cross-analysis of MIRA-chip and sequencing data indicated strong correlation between the two data sets (Supplemental Fig. S5B; Supplemental Table S8). We observed similar numbers of gain- and loss-of-methylation events on activation in the older J and S Alu subfamilies, but loss-of-methylation events dominated at AluYs (Fig. 5B), the youngest class of Alu elements. Roughly 10% of elements within the AluY family had methylation changes between naive and GC B cells, totaling over 14,000 elements (Supplemental Table S6). Validation of differential methylation status in naive and GC B cells was performed in a panel of Alu elements by genomic bisulfite sequencing (Fig. 5C; Supplemental Table S9). The distinct methylation dynamics within the AluY family suggest that either these events play a functional role in B-cell activation or that they reflect a global change in the nucleus occurring at this stage transition. Cross-analysis with ENCODE DNase I-seq data indicated that a substantially lower percentage of differentially methylated Alus, compared to DMRs, overlapped regulatory factor binding sites (Supplemental Fig. S3C). While the genomic targets of retinoic acid receptors (RARs) are frequently found in Alu elements (Laperriere et al. 2007), the presence of the RAR motif was not enriched in differentially methylated Alus in GC B cells (Supplemental Fig. S3D), further suggesting that differential methylation of these elements plays a biological role distinct from modulation of TF binding.
Similar to all DMRs between naive and GC B cells, gain-of-methylation AluYs were found to be enriched near promoter-proximal regions and in transcription units (Fig. 6A). In contrast, loss-of-methylation AluYs were spread throughout the genome, mirroring the distribution of all elements within the AluY subfamily (Fig. 6A). Chromosome distribution analysis indicated loss-of-methylation events at AluYs were global and pervasive across the genome in GC B cells upon immune activation (Fig. 6B). Nonetheless, pathway analysis of genes associated with loss-of-methylation Alus indicated enrichment of genes with functional roles in cell growth and proliferation (Supplemental Table S10), a critical component of the GC reaction. Interestingly, while gain-of-methylation distributes evenly across the entire consensus Alu element in both Y and S subfamilies, loss-of-methylation events preferentially occurred at the 5′ and 3′ ends of the elements (Figs. 6C; Supplemental Fig. S5C,D). Since these regions have well-positioned nucleosomes (Englander et al. 1993; Englander and Howard 1995), the observed 5′ and 3′ loss-of-methylation regions could reflect repositioning of nucleosomes at Alu elements in naive to GC B-cell transition. The widespread nature of Alu demethylation may therefore be indicative of a global restructuring of chromosomal architecture and nucleosome positioning in B cells upon immune activation.
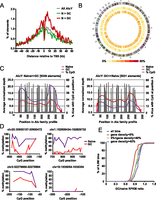
Distribution of differentially methylated Alus across the genome. (A) Distance of differentially methylated AluYs to the nearest TSS. The distribution of all AluY (dashed purple line) and both types of differentially methylated Alus, loss-of-methylation (N > GC) and gain-of-methylation (N < GC), are shown. (B) Circos plot (Krzywinski et al. 2009) illustrating the density of differentially methylated AluY elements (middle ring, GC > naive; inner ring, naive > GC) on each chromosome. Color density (from yellow to red) represents the percentage of AluY elements (from 0% to 20%) in each 5-Mb bin that is differentially methylated (in the given direction). (C) Average normalized depth of mapped reads from MIRA-seq across a consensus profile for AluY; the position in base pair is shown on the x-axis. Percentage of AluYs with CpG at each position is also shown. (Left) The average depth of AluYs that have lost methylation in GC B cells; (right) the average depth of AluYs that have gained methylation in GC B cells compared to naive B cells. (D) Analysis of methylation status of genomic regions neighboring differentially methylated Alus by genomic bisulfite sequencing. Regions proximal to four differentially methylated Alus in Figure 5C were analyzed. After bisulfite treatment and PCR amplification, at least eight individual clones of the PCR product were analyzed. The percentage of methylated cytosine (y-axis) was calculated at individual CpG positions relative to the 5′ end of each Alu element (x-axis). The genomic locations (hg19) of the Alu elements analyzed are listed above each graph. Methylation status of CpGs within individual PCR products are shown for five Alu elements in Supplemental Figure S4. (E) Overall DNA methylation dynamics at chromosomal regions with varying degrees of gene density between naive and GC B cells. We analyzed 5759 genomic bins of 0.5M bp for GC/naive adjusted RPKM ratios (there are a total of 6146 bins in the genome, but only those bins with GC content between 30% and 60% were analyzed). Gene density for a bin is defined as the fraction of positions that are within at least one RefSeq gene body. The data were partitioned into regions with low gene density (gene density < 5%, N = 1137, red line), intermediate gene density (5% < gene density < 40%, N = 1533, green line), and high gene density (gene density > 40%, N = 3100, purple line).
The genomic distribution and pervasiveness of differentially methylated AluYs prompted us to investigate whether the methylation dynamics at Alu elements reflect a global change in DNA methylation. Bisulfite sequencing was performed in naive and GC B cells at a limited number of regions proximal to differentially methylated Alus, indicating that these neighboring regions were also undergoing methylation changes (Fig. 6D; Supplemental Table S11). Since MIRA enrichment preferentially captures CpG-rich genomic regions (Supplemental Table S7), future whole-genome bisulfite sequencing in these cell types will more precisely reveal the extent of methylation change outside of Alu elements and at elements with lower CpG density.
Analysis of DNA methylation content in genomic regions varying in gene density revealed an overall decreased methylation level upon B-cell activation in genomic regions with low gene density, while regions with high gene density conversely have increased methylation content (Fig. 6E). Since we did not observe a decrease in total 5mC content, this result indicates that there is a global redistribution of CpG methylation events at gene-rich vs. gene-poor regions. These data provide further support for a global restructuring of chromosomal architecture in B cells upon activation. Collectively, the DNA methylation dynamics described here provide a useful platform to gain insights into the transcriptional network in a GC reaction, as well as uncovering potential novel regulatory elements in the genome during an immune response.
Discussion
Upon immune activation, DNA methylation in B cells is reprogrammed in a dramatic fashion at the GC stage and is not restored on differentiation to a memory or PC fate (Fig. 2B). The resulting similarity in DNA methylation patterns between the two post-GC cell types is unexpected, since the two cell types have distinct transcriptional profiles (Fig. 3). These results contrast to other differentiation models, where divergent cell fates derived from a multipotent/pluripotent progenitor adopt different epigenetic patterns (Mikkelsen et al. 2007; Ji et al. 2010). It is possible that a common methylation signature between memory and PCs partially explains the more rapid PC differentiation from memory B cells upon subsequent rechallenge with the same antigen. Our observations in the B-cell compartment indicate that both B and T cells (Messi et al. 2003; Kersh et al. 2006) employ epigenetic mechanisms in generating effective memory responses.
While it is thought that repetitive elements are constitutively methylated in somatic cells, our results unexpectedly suggest that Alu elements display differential methylation patterns during immune activation within individuals (Figs. 3, 5). Our analysis also indicated enrichment of several other types of repeats by MIRA; however, we could not determine whether they are differentially methylated in B-cell subsets due to poor mapability at unique genomic locations (data not shown). A substantial portion of the cell-type–specific differential methylation of Alus documented here correlates with decreased expression of the de novo methyltransferase DNMT3A at the GC stage (Fig. 1C,D), consistent with a model delineated by Jones and colleagues (Jeong et al. 2009). Diminished expression of DNMT3A, coupled with increased replicative demand could lead to replication-dependent loss of methylation at repetitive elements. Further, the pattern of methylation loss at Alu elements is suggestive of an alteration in nucleosome positioning at the 5′ and 3′ ends of these elements, consistent with a global alteration in chromatin architecture. It is also conceivable that the DNA methylation changes observed here reflect a shift from homogenous to heterogeneous methylation patterning as previously described (Xie et al. 2011).
The sheer number of significant changes in DNA methylation at Alus (14,000 individual AluY elements) predicts that differential methylation in repetitive elements may contribute to differential gene expression within human populations in a manner analogous to that observed previously in animal models (Bultman et al. 1994). In addition, it is likely that a subset of these methylation changes reflects or contributes to global changes in nuclear architecture associated with the acquisition of a multipotent phenotype. It will be essential to determine whether the methylation dynamics at Alu elements result in changes to their transcriptional status (Hellmann-Blumberg et al. 1993), as Alu transcripts have the capacity to modulate the global gene expression program through multiple mechanisms (Ponicsan et al. 2010).
The GC B cell is a key intermediate in T-cell–dependent humoral immune responses. Like stem or progenitor cell populations, it is self-renewing, requires a specialized biological niche and signaling environment, and ultimately results in the formation of multiple, distinct cell types (Weissman 2000). Widespread loss of DNA methylation upon immune activation resembles the epigenetic reprogramming events that occur during the generation of induced pluripotent stem (iPS) cells from somatic cells (Wernig et al. 2007; Mikkelsen et al. 2008; Kim et al.2010). DNA demethylation during this reprogramming process is a prerequisite to achieve pluripotency status; inefficient DNA demethylation results in a partially de-differentiated state (Mikkelsen et al. 2008). Takahashi and Yamanaka (2006) elegantly demonstrated that the spectrum of transcriptional and epigenetic reprogramming required to generate pluripotent cells from adult precursors could be induced by specific TFs. We propose that the loss of DNA methylation we have described during the naive to GC B-cell transition permits GC B cells to acquire the potential to differentiate toward memory or PC fates, and to generate the differential response to antigenic challenge inherent to memory cell populations.
Methods
Isolation of tonsillar B cells
FACS purification of the B-cell subset was performed as described in Lai et al. (2010). Relevant clinical data of tonsil samples are included in Supplemental Table S8. The following antibodies were used: PE-IgD (BD Pharmingen), APC-CD38 (eBioscience, HIT2), and FITC-CD20 (eBioscience, 2H7). Propidium idodide was use to exclude dead cells. Sorting was performed using the BD FACS Aria II at the NIEHS Flow Cytometry Center.
Nucleic acid extraction and manipulation
Genomic DNA and total RNA were copurified from purified cell types using the DNA/RNA Isolation Kit (Qiagen). RT-PCR was performed as described in Lai et al. (2010). For genomic bisulfite sequencing, the EZ DNA Methylation Gold Kit (Zymos) was used for bisulfite conversion of genomic DNA isolated from B-cell subsets. After PCR amplification, PCR products were cloned onto TOPO-XL vectors (Invitrogen) for subsequent sequencing.
Immunohistochemistry and immunofluorescence
Immunohistochemical and immunofluorescence staining of human tonsil sections were performed as described in Fujita et al. (2004) and Jaye et al. (2007). Primary antibodies used are as follows: DNMT1 (Novus Biologicals, NB100-264), DNMT3A (Santa Cruz Biotechnology, H295), BCL6 (Dako, PG-B6p), DNMT3B (IMEGENEX, IMG-184A), MTA3 (Fujita et al. 2004), 5mC (Eurogentec, 33D3), and 5hmC (Active Motif, 59.1)
Gene expression microarray
Fifty nanograms of total RNA from B-cell subsets was used for cDNA synthesis and amplification using the WTA2 kit (Sigma). Amplified cDNA was labeled with Cy3 random nonomers (TriLink BioTechnologies) and hybridized onto NimbleGen Gene Expression 12x135K microarrays following the manufacturer's protocol (454 Life Sciences [Roche]). The microarray slides were scanned using the Agilent G2565BA DNA Microarray Scanner. Images were processed using the NimbleScan software. A three-step robust multi-array average (RMA) normalization (Ding et al. 2008) and batch correction (Johnson and Li 2007) were performed on the data set. The RefSeq gene level expression data sets were generated by averaging expression signal from probes that overlap transcription start and end sites. Differentially expressed genes were defined using the criteria ANOVA P < 0.01 and fold change > 3.
Global DNA methylation analysis
MIRA-chip, data normalization, and identification of methylation peaks were performed as described in Lai et al. (2010). NimbleGen 2.1M Deluxe Human Promoter Arrays (454 Life Sciences [Roche]) were used. Microarray probes are between 50 and 75 bp in length. Detailed methods for peak calling, identification of DMRs, analysis of average methylation signals at genomic features, GSEA, and TF motif analysis are included in Supplemental Methods. For MIRA-seq, 1 μg of genomic DNA from purified naive or GC B cells was used to perform MIRA for library preparation. End repair, A-tailing, and adapter ligation steps were performed following Illumina's protocol. Adapter ligated DNA was size-selected for 250–600 bp on a 3% NuSieve 3:1 Agarose gel. Gel purified DNA (Qiagen) was then treated with sodium bisulfite for 5 h (EZ DNA Methylation-Gold Kit, Zymo Research), followed by 18 cycles of PCR amplification using Pfu Turbo Cx (Stratagene). The libraries were sequenced on a Genome Analyzer IIx (Illumina) as paired-end 76mers; three lanes were run for each sample. Details of bioinformatics analysis at Alu elements are described in Supplemental Methods.
Data access
Microarray expression and DNA methylation data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE24919. The MIRA-seq data have been submitted to the NCBI GEO under accession number GSE42386.
Acknowledgments
We thank the members of the Wade laboratory, Drs. T. Archer, K. Adelman, R. Jothi, J. Taylor, and M. Resnick for critical comments and suggestions for this manuscript. We thank the NIEHS Microarray, Flow Cytometry, and DNA Sequencing core facilities, as well as the NIH Intramural Sequencing Center, for their assistance. We also thank Diane Lawson for technical assistance with immunohistochemistry, Hrisavgi Kondilis-Mangum for assistance with graphics, and Dr. M. Kondo for assistance with cell sorting. This work was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH (project number Z01ES101965 to P.A.W.), and by grants from the National Institutes of Health (DK60647 to D.L.J.). D.M., D.P., and R.S. were supported in whole or in part with federal funds from the National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, under delivery order number HHSN291200555547C and GSA contract number GS-00F-003L.
Footnotes
-
↵7 Corresponding author
E-mail wadep2{at}niehs.nih.gov
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155473.113.
- Received January 28, 2013.
- Accepted August 29, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








