
Massively parallel synthetic promoter assays reveal the in vivo effects of binding site variants
- Center for Genome Sciences and Systems Biology, Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63108, USA
-
↵1 These authors contributed equally to this work.
Abstract
Gene promoters typically contain multiple transcription factor binding sites (TFBSs), which may vary in affinity for their cognate transcription factors (TFs). One major challenge in studying cis-regulation is to understand how TFBS variants affect gene expression. We studied the in vivo effects of TFBS variants on cis-regulation using synthetic promoters coupled with a thermodynamic model of TF binding. We measured expression driven by each promoter with RNA-seq of transcribed sequence barcodes. This allowed reporter genes to be highly multiplexed and increased our statistical power to detect the effects of TFBS variants. We analyzed the effects of TFBS variants using a thermodynamic framework that models both TF-DNA interactions and TF-TF interactions. We found that this system accurately estimates the in vivo relative affinities of TFBSs and predicts unexpected interactions between several TFBSs. Our results reveal that binding site variants can have complex effects on gene expression due to differences in TFBS affinity for cognate TFs and differences in TFBS specificity for noncognate TFs.
Transcription factors (TFs) orchestrate programs of gene expression by binding promoters and interacting with the core transcriptional machinery. Promoters typically contain multiple transcription factor binding sites (TFBSs) with varying affinities for their cognate TFs. Analyses of TFBS variants must account for the effects of low-affinity sites, which often have important and surprising roles in gene regulation, especially when TFs bind cooperatively (Driever et al. 1989; Jiang and Levine 1993; Wharton et al. 2004; Gertz et al. 2009; Parker et al. 2011; Peterson et al. 2012). Position weight matrix (PWM) models (Stormo 2000) of binding affinities facilitate the study of TFBS variants; however, these models are often developed in vitro and offer a limited picture of the in vivo effects of variants on gene expression. The effect of a TFBS variant on gene expression is a function of the sum of its effects on binding by, potentially, all other TFs present in the nucleus. In support of this model, recent genome-wide binding studies show a striking overlap of TF binding profiles (Neph et al. 2012). Therefore, given all the possible interactions between TFs and between TFs and DNA, it is difficult to model and predict the in vivo effects of TFBS variants. The analysis of TFBS variants is particularly relevant in light of studies of human genetic variation (The 1000 Genomes Project Consortium 2012) and the role of noncoding polymorphisms in complex traits and disease (Degner et al. 2012; Maurano et al. 2012). Progress in this field requires methods to study the effects of combinations of TFBS variants inside cells.
Synthetic promoters are powerful tools for studying cis-regulation (Cox et al. 2007; Gertz and Cohen 2009; Gertz et al. 2009; Mogno et al. 2010; Raveh-Sadka et al. 2012; Sharon et al. 2012). Recent advances in DNA synthesis and high-throughput sequencing have driven the development of novel techniques for measuring large numbers of synthetic promoters (Kwasnieski et al. 2012; Melnikov et al. 2012; Patwardhan et al. 2012; Sharon et al. 2012; Arnold et al. 2013). These techniques add transcribed sequence barcodes to traditional fluorescent reporter genes, allowing reporter genes to be highly multiplexed and assayed by RNA-seq. To date, all of these methods rely on plasmid-based reporter gene libraries. Limitations in the length of synthesized DNA restrict some of these techniques to assaying relatively short synthetic regulatory elements. Here we present a method to assay large numbers of chromosomally integrated synthetic promoters of arbitrary size. We implemented the method in the yeast Saccharomyces cerevisiae and used it to study the effects of TFBS variants on cis-regulation.
The method we developed is a variant of CRE-seq (Kwasnieski et al. 2012), a technique created to transiently assay large numbers of cis-regulatory elements in mammalian cells. The modifications we made to this technique allow us to sample large numbers of chromosomally integrated synthetic promoters consisting of combinations of TFBSs with differing affinity. This large sampling was necessary to obtain the statistical power necessary to model the effects of TFBS variants on cis-regulation. We fit a thermodynamic model to the resulting data, which provides a formal framework to describe the system in terms of TF binding to DNA, and interactions between TFs. We found that binding site variants have complex effects on gene expression that are due to both differences in affinity for their cognate TFs, and differences in specificity for noncognate TFs.
Results
Construction of a barcoded synthetic promoter library
We sought to understand how sequence variants of TFBSs affect gene expression. We previously used libraries of fluorescent reporter genes to study cis-regulatory interactions between four TFBSs, which correspond to binding sites for Mig1, Reb1, Rap1, and Gcr1 (Gertz et al. 2009; Mogno et al. 2010). To build on our previous results with consensus TFBSs, we chose to create libraries consisting of variants of these same four sites. For each of the four TFs, we chose three variants, with differing predicted affinity, for a total of twelve TFBSs (e.g., Mig11, Mig12, and Mig13 denote three variants of the Mig1 TFBS). Table 1 shows the specific sequences we chose and the estimated affinities to their cognate TFs as calculated with a position weight matrix (PWM) model (Stormo and Fields 1998; MacIsaac et al. 2006) based on ChIP data (Harbison et al. 2004). We tested TFBSs with a wide variety of predicted affinities, from very high (Mig11), to very low predicted affinity (Reb13 and Rap12). Because the increase from four to twelve TFBSs entails an exponential increase of the number of possible synthetic cis-regulatory elements (CREs), we implemented CRE-seq technology to multiplex our expression measurements.
Twelve TFBS sequences in our library, including S. cerevisiae promoters where they are present and the PWM score (MacIsaac et al. 2006)
We built a CRE-seq reporter library in which each synthetic CRE reporter gene contained a unique sequence barcode (BC) in its 3′ UTR. We first synthesized double-stranded oligonucleotides (oligos) corresponding to each of the three TFBS variants for each TF: Mig1, Reb1, Rap1, and Gcr1 (Table 1). These oligos were pooled and then randomly ligated to form a library of synthetic CREs (Fig. 1A; Supplemental Fig. S1), which was cloned into a plasmid. We then inserted a library of random 15-nucleotide (nt) barcodes downstream from the CREs, such that each barcode uniquely identified a specific CRE. We performed this cloning step in such a way that each CRE was attached to more than one unique barcode: Our final library contained 7289 barcodes representing 2534 unique CREs (Supplemental Fig. S2). This redundancy increases our statistical power by providing multiple expression readouts for any specific CRE.
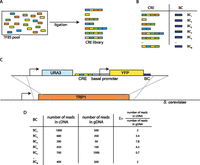
Schematic of the CRE-seq method adapted for this study. (A) Double-stranded oligonucleotides encoding TFBS are mixed in a pool and ligated randomly to create a CRE library. (B) After cloning CRE and barcode (BC) sequences into a reporter plasmid, the concordance between CREs and BCs is determined with a paired-end next-generation sequencing run. Each BC identifies a single CRE. (C) The cassette containing the library of CREs upstream of a basal promoter driving YFP and BC is integrated into the S. cerevisiae genome at the TRP1 locus by selecting for URA+ cells. (D) Cells are grown in liquid culture, and gDNA and RNA are harvested. The fraction of reads in the cDNA pool divided by the fraction of reads in the gDNA pool for each BC is a quantitative measurement of the expression driven by the corresponding CRE.
We matched the barcodes to their specific CRE using paired-end sequencing of the plasmid library containing the CREs and barcodes (Fig. 1B; Supplemental Fig. S1; Supplemental Data 1), coupled with a naïve clustering algorithm (Methods). We were careful not to use PCR to prepare the library for sequencing, as we found that PCR amplification creates chimeric products that scramble the CRE-barcode associations. After determining the CRE-barcode associations, we cloned a cassette containing a basal promoter (TSA1) driving yellow fluorescent protein (YFP) into the library, between the CREs and the barcodes. The entire library cassette was then excised and inserted into the S. cerevisiae genome at the TRP1 locus (Fig. 1C).
To measure, in parallel, the expression driven by each CRE, we grew the integrated yeast library and then sequenced the barcodes (Supplemental Data 2) from harvested RNA and genomic DNA (gDNA). We computed the cDNA/gDNA ratio of each barcode and used the median ratio for all barcodes corresponding to a particular CRE as the expression of that CRE (Supplemental Data 3).
CRE-seq accurately measures gene expression
To test the accuracy of the CRE-seq method in S. cerevisiae, we compared expression measurements made by CRE-seq to those made by flow cytometry. We picked 337 CREs containing sites for Mig11, Reb11, Rap11, and Gcr11 and measured their expression in glucose minimal media by flow cytometry. We then pooled all strains and measured their expression in glucose minimal media by CRE-seq. The high correlation (r = 0.92) between pooled CRE-seq measurements and individual flow cytometer measurements confirms that CRE-seq accurately measures gene expression in our system (Fig. 2A).
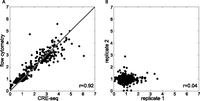
CRE-seq accurately measures gene expression. (A) Comparison between expression measured by CRE-seq and flow cytometry. Each dot represents a CRE whose activity has been measured with a traditional fluorescent assay (y-axis) and with CRE-seq (x-axis). The high correlation indicates that CRE-seq expression measurements are as accurate as those measured by traditional fluorescent assay. The line represents the perfect model (r = 1). (B) Biological replicates of a CRE-seq library where expression is controlled by one CRE matched to 602 different BCs. The library was grown and harvested two times; CRE-seq was performed independently on each replicate. Replicate measurements of BC expression are plotted on the x-axis (replicate 1) and on the y-axis (replicate 2). The absence of correlation reveals that the BCs have no reproducible effects on gene expression.
To verify that the 15-bp barcodes in the 3′ UTR of the reporter genes do not affect our measurements, we assayed the effects of barcode sequences on expression. Using CRE-seq, we assayed the expression of 602 clones of the same promoter, in which each clone contained a different barcode sequence in its 3′ UTR. We performed two replicates of this experiment. If the barcodes had an effect on gene expression, we would expect to see a positive correlation between the two replicates, as barcodes that increased reporter expression would be correlated between replicates. However, we observed a low correlation between the two replicates (r = 0.04). The lack of correlation demonstrates that the random barcodes in the 3′ UTR do not have reproducible effects on expression (Fig. 2B).
Model selection
After verifying the accuracy of our assay, we analyzed the full library, composed of 7289 BCs for 2534 CREs. To understand the rules of combinatorial regulation, we applied a thermodynamic model to our data. This model is a formal framework that describes the data in terms of TF binding to DNA and interactions between TFs, and provides an automated method to detect the effects of TFBS in large sets of promoters (Gertz and Cohen 2009; Gertz et al. 2009; Mogno et al. 2010). Because the differences in expression between members of our library were not correlated with predicted nucleosome occupancy (R2 = 0.033), we did not explicitly model interactions with nucleosomes (Kaplan et al. 2009).
We first analyzed CREs containing only Mig11, Reb11, Rap11, and Gcr11 sites and recapitulated the results (Supplemental Table S1; Gertz et al. 2009; Mogno et al. 2010) , showing that Mig11 sites act cooperatively to repress expression, while the Reb11, Rap11, and Gcr11 sites all have activating effects. We, therefore, demonstrated that the trends in expression data from CRE-seq recapitulate the trends in expression measured by traditional reporter gene assays.
To explore different potential mechanisms that could account for the effects of TFBS variants, we applied several thermodynamic architectures to the full data set with all 12 TFBSs. We started with the simplest set of hypotheses: Each TF binds at its three cognate TFBSs with different affinities (Fig. 3A). We also included a parameter to represent the Mig1-Mig1 cooperative interaction that was found in Gertz et al. (2009) and verified in our data.
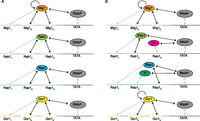
The thermodynamic model consists of a set of interactions that govern TF-DNA and TF-TF binding. Each arrow represents an interaction included in the model in the form of a parameter proportional to the ΔG. Black arrows represent the free parameters. (A) The set of interactions allowed in the simplest model: Each TF is allowed to bind to its cognate TFBSs and to interact with polymerase. Mig1 is allowed to interact with itself when two or more Mig1 sites are present in the same promoter. (B) The set of interactions applied to the model with the highest score: A protein X other than Reb1 is allowed to bind at site Reb13, and a protein Y other than Rap1 is allowed to bind at site Rap12. Both Mig1 and Gcr1 are allowed to interact with themselves when two or more of their sites are present in the same promoter.
We then asked whether our data supported a model with additional interactions. We started by generating a list of additional features (hypotheses) that were not present in the initial simple model, including (1) an interaction term between Rap1 and Gcr1, as suggested by Scott and Baker (1993) and Tornow et al. (1993), (2) a cooperativity term for Gcr1, as suggested by our expression data (Supplemental Fig. S3) and by Scott and Baker (1993), (3) a term allowing a protein (X) other than Reb1 to bind the Reb13 site, and (4) a term allowing a protein (Y) other than Rap1 to bind the Rap12 site. We included features 3 and 4 because Reb13 and Rap12 had strong effects on expression, even though PWM analysis of these sites indicates that they have very low affinities for their TFs, which suggests that their effects may be mediated through the binding of other TFs.
After identifying a set of features that might improve the simple model, we constructed several model architectures including these additional features in various combinations. Each model was fit to the measured expression values and scored based on the sum of squares of the residuals (RSS) and the number of free parameters needed for the fit, introducing a greater penalty for models with more free parameters. When we rank our models based on this score, a clear pattern appears (Fig. 4A): the addition of the Rap1-Gcr1 interaction consistently lowers the model score (worse model), while adding Gcr1 cooperativity always results in a higher score (better model). Moreover, allowing unknown proteins to bind the Reb13 and Rap12 sites (six TFs in total) results in a better model even after penalization for increased parameter number. The best performing model includes parameters representing Mig1 self-cooperativity, Gcr1 self-cooperativity, a protein (X) other than Reb1 binding at site Reb13, and a protein (Y) other that Rap1 binding at site Rap12 (Fig. 3B). Scoring Reb13 and Rap12 against PWMs for known TFs (Spivak and Stormo 2012) suggests that Reb13 may be bound by Rtg1 (P = 0.0032) and that Rap12 may be bound by Yer130C (P = 0.0059). This result is not surprising given that the PWM models for these two sites (Reb13 and Rap12) predict extremely low affinity for their cognate TFs (see Table 1). It is, therefore, reasonable to expect other TFs to bind to these particular sites.
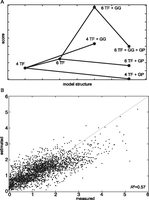
(A) Several model structures with different sets of rules have been applied to the data. Each dot represents a specific model, whose score is on the y-axis. The score is plotted as the absolute value of the AIC score, calculated taking into account the RSS and a penalty term for the number of free parameters (Methods). An increase in the plotted score (thus a decrease in the AIC score) indicates a better model. Increasing the number of TFs included in the model from four to six increases the score. The addition of the Gcr1-Gcr1 (GG) interaction always results in a better score. The addition of the Gcr1-Rap1 (GP) interaction always results in a worse score. The model with the best score is the one with six TFs and the Gcr1-Gcr1 interaction. All models represented in this plot include the Mig1-Mig1 interaction. (B) The thermodynamic model with six TFs and Gcr1-Gcr1 interaction accurately predicts synthetic promoter gene expression. Each dot represents expression driven by one of the 2534 CREs we assayed in this study. The measured CRE-seq expression is on the x-axis, while the predicted expression from the thermodynamic model is shown on the y-axis. R2 = 0.57 shows that our model explains 57% of the variance in the data. The line represents the perfect model (R2 = 1).
This model explains 57% (P << 0.01) of all the variance in expression in our 12-site synthetic promoter library (Fig. 4B). For each model, we performed 100 independent fits. In general, model fits converged 40% of the time, and these parameters were within the 95% confidence interval of the solution. We performed repeated random subsampling validation (Supplemental Fig. S5), showing that we obtain similar results with ∼1000 unique promoters. However, to obtain reliable estimates of some parameters, at least 2000 unique promoters are necessary. Thus, the extra statistical power afforded by CRE-seq allowed us to identify features of this system that were undetectable in our previous experiments with smaller libraries (Gertz and Cohen 2009; Gertz et al. 2009; Mogno et al. 2010).
The thermodynamic model predicts in vivo relative affinities between TFs and DNA
We next asked whether the in vivo predicted affinities estimated from our thermodynamic model match the PWM predictions from ChIP-seq data. We computed ΔΔG for pairs of binding sites. A negative ΔΔG indicates a stronger TFBS (with higher affinity), while a positive ΔΔG indicates a weaker site. When the PWM and our thermodynamic model are in agreement, the ΔΔG calculated with the PWM and the ΔΔG calculated with the thermodynamic model are proportional. For example, our model is in good agreement with the PWM model for Mig1 (Table 2, rows 1 and 2). Our model also agrees with PWM predictions that Rap13 is stronger than Rap11 (Table 2, row 3). In contrast, our model predicts Reb12 to be stronger than Reb11, while the PWM model predicts the opposite (Table 2, row 4).
Comparison between TFBS affinities predicted by thermodynamic modeling and PWM analysis
Our relative affinity predictions for Gcr1 do not agree with PWM models (Table 2, rows 5 and 6). Our model predicts that Gcr12 is the strongest site for Gcr1, while the PWM model predicts Gcr13 to be the strongest site. The PWM model was generated using genome-wide chromatin immunoprecipitation (ChIP) data collected in a rich media (Harbison et al. 2004; MacIsaac et al. 2006); our predictions are estimated from measuring synthetic promoter expression in minimal media. It is possible that the inconsistencies in these predictions can be explained by differences in growing conditions or by differences between measuring binding versus activity through a gene expression-based reporter assay. We also tried using different PWM models for Gcr1, which came from different experiments. None of these PWMs for Gcr1 are in good agreement with each other, nor do they agree well with our predictions (Harbison et al. 2004; MacIsaac et al. 2006; Pachkov et al. 2007; Foat et al. 2008; Spivak and Stormo 2012). The relationship between occupancy at the Gcr1 sites and the sites' effect on gene expression may be complicated by condition-specific binding of Gcr1 or the binding of other factors. In the following analysis, we refer to Gcr12 as the strongest site and Gcr11 as the weakest site, as predicted by our model.
Gcr1 participates in complex TF-TF interactions
The Gcr1 binding sites used in this study showed the ability to enhance the activity of surrounding TFBS, regardless of whether activators or repressors bind to those TFBSs. When Gcr11 sites are added to promoters containing only Mig11 sites, their average expression decreases (Fig. 5A). In contrast, when Gcr11 sites are added to promoters containing only Reb11 or Rap11 sites, the expression of the reporter gene increases. We also observe a similar behavior when Gcr11 is added to Reb12, Reb13, Rap12, and Rap13. However, the ability of Gcr11 to repress is weaker when it is coupled with weaker sites for Mig1 (e.g., Mig12 and Mig13) (Fig. 5B). The data suggest that the Gcr11 site acts as an activator when next to any activator site, but it acts as a repressor when next to a strong Mig1 site, and has little effect next to a weak Mig1 site. Increasing the predicted affinity of the Gcr1 site hides this behavior: Gcr13, a stronger site, has a smaller effect on the Mig11 site (Fig. 5C). The repressing effect disappears when we use Gcr12, the highest affinity site. Moreover, this effect is particularly strong in promoters in which Gcr11 and Mig11 sites are adjacent to each other (Supplemental Fig. S4). These data seem to suggest a role of the Gcr1 sites in facilitating the binding of other TFs and increasing their regulatory potential.
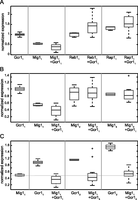
Gcr1 binding sites have complex effects on expression. (A) When Gcr11 sites are added to promoters containing only Reb11 or Rap11 TFBSs, their effect is to increase the activation of gene expression. However, when Gcr11 sites are added to promoters containing only Mig11 sites, their effect on gene expression is repressive. (B) Gcr11 TFBS has a weaker repressive effect when added to low-affinity Mig1 sites (Mig12 and Mig13). (C) Gcr1 TFBS with low affinity (Gcr13 and Gcr12) have weak repressive interactions when combined with high-affinity Mig1 sites (Mig11).
Discussion
We adapted CRE-seq for use with synthetic promoters of arbitrary size integrated into the genome of S. cerevisiae. With the development of CRE-seq, we can assay thousands of integrated synthetic promoters, a 10-fold increase over what was previously possible with fluorescent reporter genes. We showed that the method is accurate and reproducible and that the barcodes in the 3′ UTR of the reporter gene do not affect gene expression. As technologies for genome editing (Christian et al. 2010; Bogdanove and Voytas 2011) become more efficient, we anticipate using CRE-seq to study synthetic promoters integrated into the genomes of mammalian cells.
An advantage of CRE-seq is that it allows us to build larger libraries since all clones are built and assayed in parallel. It also overcomes some of the limitations of traditional assays based on flow cytometery, such as limited dynamic range. CRE-seq measures the abundance of mRNA rather than stable fluorescent proteins, whose long half-lives could mask the true promoter activity.
We used CRE-seq to obtain the statistical power necessary to study cis-regulation in promoters containing combinations of TFBS variants. The increased power we obtained from analyzing large libraries revealed TFBS effects that we could not detect in smaller libraries composed of the same binding sites. This demonstrates the utility of CRE-seq when applied to synthetic promoters. In many cases, our binding affinity predictions agree well with established PWM models of binding (MacIsaac et al. 2006). In cases where our predictions were discordant with PWM predictions, as was the case for Reb13 and Rap12, we found that our data supported a model in which these variant TFBSs are recognized by other TFs. We think the differences in these predictions stem from different experimental conditions and the fact that in vitro binding is not equivalent to in vivo expression potential.
Our work uncovered an unusual interaction between Gcr1 and Mig1. Although Gcr1 sites behave as weak activators, when put in combination with repressive Mig1 sites, Gcr1 sites increase the repressive effects of Mig1. One possible explanation is that Gcr1 binding opens the locus, thus facilitating the binding of Mig1. This manifests as a greater repressive effect of Mig1 but only when the activating potential of Gcr1 is weak.
With the increasing power and affordability of next-generation sequencing technologies, we anticipate that CRE-seq will be a useful tool for unraveling other kinds of interactions between cis-regulatory sequences.
Methods
Construction of the CRE-BC library
Escherichia coli strain DH5α was used for all bacterial cloning steps. Plasmid pIM202 was derived from pIM102 (Mogno et al. 2010) by removing the TSA1 promoter-YFP cassette and replacing it with a multiple cloning site (containing sites for BglII, XmaI, BamHI, KpnI, ClaI, EagI, AvrII, and XbaI restriction enzymes). CREs were cloned into pIM202 at the BamHI site as in Gertz and Cohen (2009), Gertz et al. (2009), and Mogno et al. (2010), and ∼7000 colonies were scraped for DNA extraction using a maxi-prep kit (Sigma GenElute HP Plasmid Maxiprep Kit).
To create random barcodes (BCs), two oligos containing 15 random nucleotides flanked by 6 or 7 bases (oligos prIM01 and prIM02) (Supplemental Table S2) were denatured at 95°C in a water bath and then annealed for 16 h until the water reached room temperature. The BCs were then cloned into the CRE plasmid library using restriction sites EagI HF and XbaI. The ligations were digested with AvrII before transformation to reduce background. Roughly 20,000 colonies were then scraped and maxi-prepped (Sigma GenElute HP Plasmid Maxiprep Kit) at this step. The TSA1 promoter-YFP cassette was amplified from plasmid pIM102 (98°C for 1 min, 5 cycles: 98°C for 15 sec, 56°C for 30 sec, 72°C for 60 sec, 25 cycles: 98°C for 15 sec, 63°C for 30 sec, 72°C for 60 sec, and 72°C for 5 min; NEB HF Phusion MM) using primers prIM03 and prIM04 (Supplemental Table S2) and cloned into the library using restriction enzymes KpnI and EagI HF. The ligation mix was digested with ClaI after ligation to reduce background. About 35,000 colonies were picked at this step. The CRE-BC plasmid library was integrated into S. cerevisiae BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) at the TRP1 locus, following the procedure described in Gertz and Cohen (2009), Gertz et al. (2009), and Mogno et al. (2010). Between 7000 and 8000 S. cerevisiae colonies were replicated onto SC media with 2% glucose and 5-FAA (5-Fluoroanthranilic acid) to enrich for the colonies carrying the correct integration. These colonies were scraped and pooled for growth and expression assays.
Matching CREs to BCs
CREs and BCs were matched after cloning the BCs into the plasmid library but before inserting the TSA1 promoter -YFP cassette. The plasmid library was digested with restriction enzymes XmaI and XbaI. Illumina paired-end adaptors were ligated, and the DNA molecules between 250 and 500 base pairs in length were selected on an agarose gel. No PCR was performed to prevent chimeric products that mask the correct CRE-BC pairs. The purified DNA was then sequenced with an Illumina MiSeq run using a paired-end 250 × 50 bp protocol to sequence the CRE and BC regions, respectively. We obtained about 1 million reads. BCs represented by fewer than five reads were not used in the analysis. Occasionally, more than one CRE was associated with a particular BC. In this case, the CRE with the highest number of reads was assigned to the BC if and only if it was represented by at least 90% of the total number of reads associated with the BC (Supplemental Data 1). Otherwise, the BC was not included in our analysis. Subsequently, all BCs associated with the same CRE were analyzed. We calculated the pairwise sequence distance for all BCs representing the same CRE, and we eliminated the ones that had similar sequences to another BC of higher rank, assuming that they arose from sequencing errors.
Flow cytometer assay
The strains used for the validation experiment (Fig. 2A), were picked from the transformation plate and arrayed into 96-well microplates. The CREs and the BCs were sequenced with a Sanger protocol (Beckman Coulter Genomics). Cultures were grown in 500 μl of synthetic complete media lacking uracil with 2% glucose with shaking at 30°C in 2-mL 96-well plates for 4 h. The cells were then fixed with paraformaldehyde as described in Gertz and Cohen (2009), Gertz et al. (2009), and Mogno et al. (2010). The fluorescence intensities and electronic volumes of 25,000 cells from each well were measured on a Beckman Coulter Cell Lab Quanta SC with a multiplate loader. Fluorescence was then divided by volume to obtain a normalized fluorescence value for every cell. For each well, mean and variance were calculated from the normalized fluorescence values for 25,000 events.
CRE-seq
The S. cerevisiae library was grown in synthetic complete media lacking uracil with 2% glucose with shaking at 30°C. After 5 h, gDNA and total RNA were harvested. RNA was then treated with TURBO DNase (Ambion) to eliminate genomic DNA contamination, and cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen), with oligo-dT primers (IDT). The 3′ UTR of the YFP gene, containing the BC, was amplified from gDNA and from cDNA (98°C for 1 min, 5 cycles: 98°C for 15 sec, 54°C for 30 sec, 72°C for 45 sec, 10 cycles: 98°C for 15 sec, 58°C for 30 sec, 72°C for 45 sec, and 72°C for 5 min; Phusion High-Fidelity PCR Master Mix [NEB]) using primers prIM05 and prIM06 (Supplemental Table S2). We also used primers that amplify across the integration region, prIM05 and prIM07 (Supplemental Table S2), on the gDNA to select for correct integrations. Only the BCs represented in this second control gDNA PCR were included in our analysis. The PCR products were purified with a QIAquick PCR Purification Kit (Qiagen), digested with EagI HF and XhoI, and ligated to Illumina adaptor sequences. The final product was amplified (98°C for 1 min, 12 cycles: 98°C for 15 sec, 63°C for 30 sec, 72°C for 45 sec, and 72°C for 5 min) with primers prIM08 and prIM09 (Supplemental Table S2) to enrich for molecules containing both adaptor sequences. This library was run on two lanes of an Illumina HiSeq machine, generating ∼102 million reads. Only barcodes with >25 reads in the gDNA pool and at least one read in the cDNA pool were used for the analysis, for a total of 7289 BCs. Expression associated with each BC was then calculated as the number of reads in the cDNA pool divided by the number of reads in the gDNA pool (for the same set of primers). These 7289 BCs mapped to 2633 unique CREs (Supplemental Data 2). Subsequently, we determined that 99 of these CREs were likely to contain mutations that altered their expression (see “Outlier detection” below). The distribution of BCs identifying each promoter was uneven; 16.1% of the promoters had at least three BCs associated with them, while the remaining 83.9% had two or one (Supplemental Fig. S2; Supplemental Data 3). Finally, expression driven by each CRE was calculated as the median ratio of all the BCs associated with it.
Thermodynamic model
To model gene expression, we implemented a thermodynamic model of polymerase occupancy originally proposed by Shea and Ackers (1985). The model and implementation were described previously in Gertz and Cohen (2009), Gertz et al. (2009), and Mogno et al. (2010), and it includes parameters proportional to ΔGs of the interactions between proteins and DNA and between proteins. We did not model nucleosome effects. We scanned our promoter sequences with the Nucleosome Positioning prediction software (Kaplan et al. 2008) and found very low correlation between predicted nucleosome occupancy averaged across the TFBS region and the measured expression (r = 0.184). Moreover, the averaged nucleosome occupancy predictions were very similar across our promoter sequences (CV = 0.06). The Akaike Information Criterion (Akaike 1974), which introduces a penalty term for the number of parameters, was used for model selection. Repeated random subsampling validation was performed for cross-validation with training sets containing 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80%, and 90% of the total number of data points. All calculations were performed using the Matlab package from The Mathworks, Inc.
Outlier detection
Given the high rate of mutations in S. cerevisiae transformants, we expect 5%–6% of the colonies to contain mutations that could affect gene expression. The CREs and the BCs were sequenced and matched before inserting the basal promoter and YFP gene, and before the transformation into S. cerevisiae; therefore, we did not detect mutations in subsequent steps. CREs represented by three or more BCs are not affected by this problem, since outlier detection is an easy task in these cases. However, our library contains 1806 CREs associated either with one BC only, or with multiple BCs, and high variance in expression (CV > 0.5). Replicate experiments showed that 95% of the CREs represented by only one BC produce an accurate measure of gene expression. Instead of eliminating all the CREs represented by a low number of BCs, we used the thermodynamic model in a recursive way to identify the true outliers.
The first step was to apply the thermodynamic model only to the 827 CREs represented by two or more BCs and characterized by low expression variance (CV < 0.5). The fit model was used to calculate the error for each of the excluded 1806 CREs. The excluded CREs were ranked based on the error and reintroduced to the model one at a time until the overall R2 dropped 10% with respect to the original model. This resulted in the exclusion of about 100 CREs. Then, the thermodynamic model was applied only to the selected CREs. The CREs excluded from our analysis represent the ones whose expression cannot be explained by the model. There could be two reasons for this: (1) They contain high measurement error; or (2) they contain a specific feature not included in the model. To test whether these CREs contain features that we were not capturing with our model, we looked at the sequence contents of these excluded CREs: They were not enriched in length (number of building blocks), and they were not enriched in any specific TFBS or in any pair of TFBSs. We also tested several models: We added parameters to include four or six TFs, and to capture the Gcr1-Gcr1 and the Gcr1-Rap1 sites interactions. Each time, we repeated this recursive procedure, excluding between 96 and 115 CREs, and found no common sequence feature in the excluded sets. Moreover, the pairwise intersections of the excluded sets were always between 96% and 100%, indicating a small, reproducible set of outliers. After these analyses, we concluded that the unexplained expression for these outlier promoters must be due either to sequencing errors or to secondary mutations that occurred during their transformation into S. cerevisiae. We excluded these outliers from our final analysis, obtaining a final set of 2534 CREs.
PWM analysis
PWM models for TF binding (MacIsaac et al. 2006; Pachkov et al. 2007; Foat et al. 2008; Spivak and Stormo 2012) were used to estimate the affinity of TFs to their cognate TFBSs. We used patser (Stormo et al. 1982) to calculate these scores. The PWM scores are proportional to the –ΔG of the interaction.
Acknowledgments
We thank Chris Fiore and Robi Mitra for discussions on the CRE-seq technical details; Aaron Spivak for assistance with PWM analysis; and Jessica Hoisington-Lopez, Henning Seedorf, Michelle Smith, Brian Muegge, and Jeffrey Gordon for next-generation sequencing support. This work was supported by a grant from the National Institutes of Health (GM092910-02).
Author contributions: I.M. and J.C.K. designed and performed the experiments, I.M. analyzed the data, B.A.C. supervised the project, and I.M., J.C.K., and B.A.C. wrote the paper.
Footnotes
-
↵2 Corresponding author
E-mail cohen{at}genetics.wustl.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157891.113.
- Received March 19, 2013.
- Accepted July 30, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








