
Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus
- Xiaoyang Zhang1,4,
- Richard Cowper-Sal·lari1,2,4,
- Swneke D. Bailey3,
- Jason H. Moore1,2 and
- Mathieu Lupien1,3,5
- 1Department of Genetics and Institute for Quantitative Biomedical Sciences, Norris Cotton Cancer Center, Dartmouth Medical School, Lebanon, New Hampshire 03756, USA;
- 2Computational Genetics Laboratory and Department of Genetics, Norris Cotton Cancer Center, Dartmouth Medical School, Lebanon, New Hampshire 03756, USA;
- 3Ontario Cancer Institute, Princess Margaret Hospital–University Health Network, and the Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada
-
↵4 These authors contributed equally to this work.
Abstract
Genome-wide association studies (GWAS) are identifying genetic predisposition to various diseases. The 17q24.3 locus harbors the single nucleotide polymorphism (SNP) rs1859962 that is statistically associated with prostate cancer (PCa). It defines a 130-kb linkage disequilibrium (LD) block that lies in an ∼2-Mb gene desert area. The functional biology driving the risk associated with this LD block is unknown. Here, we integrate genome-wide chromatin landscape data sets, namely, epigenomes and chromatin openness from diverse cell types. This identifies a PCa-specific enhancer within the rs1859962 risk LD block that establishes a 1-Mb chromatin loop with the SOX9 gene. The rs8072254 and rs1859961 SNPs mapping to this enhancer impose allele-specific gene expression. The variant allele of rs8072254 facilitates androgen receptor (AR) binding driving increased enhancer activity. The variant allele of rs1859961 decreases FOXA1 binding while increasing AP-1 binding. The latter is key to imposing allele-specific gene expression. The rs8072254 variant in strong LD with the rs1859962 risk SNP can account for the risk associated with this locus, while rs1859961 is a rare variant less likely to contribute to the risk associated with this LD block. Together, our results demonstrate that multiple genetic variants mapping to a unique enhancer looping to the SOX9 oncogene can account for the risk associated with the PCa 17q24.3 locus. Allele-specific recruitment of the transcription factors androgen receptor (AR) and activating protein-1 (AP-1) account for the increased enhancer activity ascribed to this PCa-risk LD block. This further supports the notion that an integrative genomics approach can identify the functional biology disrupted by genetic risk variants.
Whole-genome sequencing efforts have identified more than 30 million genetic variants across our genome (The 1000 Genomes Project Consortium 2010). While these include structural variants such as indels and inversions, the majority are single nucleotide polymorphisms (SNPs). Using these SNPs, genome-wide association studies (GWAS) have identified more than 4800 genetic risk variants associated with more than 500 human traits and disorders (Hindorff et al. 2009). Most of these risk variants map to poorly annotated noncoding regions (Frazer et al. 2009). This has hindered discoveries of exactly how these may impact the phenotypes under scrutiny. Recently, four independent GWAS studies performed in populations of European ancestry have shown that the SNP rs1859962 located within the 17q24.3 locus is associated with PCa susceptibility with an odds ratio higher than 1.2 (Gudmundsson et al. 2007; Eeles et al. 2008; Eeles et al. 2009; Schumacher et al. 2011). The SNP rs1859962 is located in a gene desert area, where its closest genes including SOX9, KCNJ2, and KCNJ16 are ∼1 Mb away (Gudmundsson et al. 2007). Like most of the other noncoding risk variants, the functional biology underlying this SNP is unknown.
Currently, a series of projects, including the ENCODE Project, NURSA, the Roadmap Epigenome Project, and the International Human Epigenome Project, are leading efforts to annotate features across the genome, including noncoding regions (The ENCODE Project Consortium 2007; Bernstein et al. 2010; Ernst et al. 2011; Tang et al. 2011). These benefit from approaches exploiting massively parallel sequencing technologies such as ChIP-seq, DNase I-seq, and RNA-seq (The ENCODE Project Consortium 2007; Morozova et al. 2009; Ernst et al. 2011). Through epigenomic mapping, functional regulatory elements such as enhancers, promoters, and insulators have been defined (The ENCODE Project Consortium 2007; Heintzman et al. 2007; Kim et al. 2007; Ernst et al. 2011). Transcriptome and epigenome mapping has also uncovered known as well as new transcriptional units, such as long noncoding RNA (lncRNA) (Harrow et al. 2006; Guttman et al. 2009). Finally, cistrome analysis has identified specific binding sites for a wide range of DNA binding proteins, including transcription factors (Lupien et al. 2008; Heintzman et al. 2009; Wang et al. 2009; Tang et al. 2011). These genome-wide annotation efforts, done in cell types originating from various tissues, revealed tissue-type specificities (The ENCODE Project Consortium 2007; Bernstein et al. 2010; Ernst et al. 2011; Tang et al. 2011). For instance, enhancers were shown to be distributed in a tissue-type-specific manner via epigenomic mapping in multiple human cell lines, thus resulting in tissue-type-specific transcriptional programs (Lupien et al. 2008; Heintzman et al. 2009; Ernst et al. 2011). These annotated genomic data are starting to shed light on how GWAS variants located in noncoding regions may influence clinically relevant phenotypes. Here, we have integrated GWAS and annotated genomic features to investigate the functional biology disrupted within the 17q24.3 risk locus in PCa cells.
Results
The 17q24.3 PCa risk locus harbors regulatory elements as opposed to transcripts
GWAS are performed using SNP arrays that contain at most 2 million of all approximately 30 million known SNPs (dbSNP132). The studies that identified the rs1859962 SNP as a genetic risk variant used lower-density SNP arrays containing about 500,000 SNPs. Hence, SNPs associated with diseases through GWAS are unlikely the causal genetic risk variant (Grant and Hakonarson 2008; Frazer et al. 2009). However, these risk-associated SNPs are to segregate with the underlying causal variant since they are in linkage disequilibrium (LD) (The International HapMap Consortium 2007). The International HapMap Project has defined LD blocks across the genome by profiling the segregation of about 4 million SNPs. This reveals that the PCa risk variant rs1859962 resides in a LD block within the 17q24.3 locus that spans ∼130 kb (Fig. 1A,B). This LD block appears to lie in a gene desert area since there are no RefSeq genes mapping to this region (Fig. 1A,B). One long noncoding RNA (lincRNA), LINC00600, resides in the LD block. However, its expression is undetectable in prostate tissues (Gudmundsson et al. 2007). Directed RT-PCR assays in LNCaP PCa cells also fail to detect significant levels of expression for LINC00600 (Supplemental Fig. S1). Epigenomic mapping for transcriptional units also supports this contention. For instance, the gene body of actively transcribed genes is enriched with nucleosomes trimethylated on lysine 36 of histone 3 (H3K36me3) (Barski et al. 2007; Guttman et al. 2009; Ernst et al. 2011), while their promoters are enriched with nucleosomes trimethylated on lysine 4 of histone 3 (H3K4me3) (Barski et al. 2007; Heintzman et al. 2007; Guttman et al. 2009; Ernst et al. 2011). This is highlighted in LNCaP and VCaP PCa cells (Korenchuk et al. 2001) by the highly expressed androgen receptor (AR) gene whose gene body and promoter are enriched for H3K36me3 and H3K4me3, respectively, defined in ChIP-seq assays (Supplemental Fig. S2; Yu et al. 2010). Furthermore, silenced genes can also be identified through epigenomic mapping because their promoters are enriched in nucleosomes trimethylated on lysine 9 of histone 3 (H3K9me3) (Barski et al. 2007). Noteworthy, the target LD block lacks enrichment for any of these modifications when assessed by ChIP-seq in LNCaP or VCaP PCa cells (Fig. 1C). Similarly, ChIP-seq assays in 10 additional cell lines of different tissues of origin (H1-hESC, HMEC, HSMM, HSMMtube, HUVEC, K562, NH-A, NHEK, NHLF, and Osteoblast) (The ENCODE Project Consortium 2007) failed to reveal any enrichment for H3K4me3 or H3K36me3 across the 17q24.3 locus (Fig. 1C; Supplemental Fig. S3). This suggests that the risk associated with the rs1859962 PCa LD block lies in a transcriptionally silent region and therefore does not directly target transcripts for disruption.
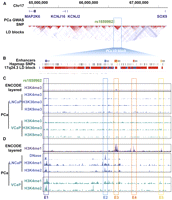
Epigenetic annotation of 17q24.3 risk locus. (A) The ∼3-Mb region surrounding the PCa GWAS SNP rs1859962 presents the annotated RefSeq genes, the HapMap LD blocks relative to the rs1859962 SNP. (B) A focused view of the rs1859962 LD block presenting regulatory elements, SNPs covered by the HapMap project, and the LD score is presented. LD score: (dark red) LOD = 2, D′ = 1; (light red) LOD = 2, D′ < 1; (blue) LOD < 2, D′ = 1; (white) LOD < 2. D′ < 1. (C) The H3K4me3 enrichment from the ENCODE Project (layered for multiple cell lines) and PCa cells (LNCaP and VCaP) as well as the H3K36me3 and H3K9me3 enrichment from PCa cells (LNCaP and VCaP) are presented across the 17q24.3 PCa risk LD block. (D) DNase I-seq enrichment from LNCaP cells, H3K4me1 enrichment from the ENCODE Project (layered for multiple cell lines) and PCa cells (LNCaP and VCaP), and H3K4me2 enrichment from PCa cells (LNCaP and VCaP) are indicated. (Colored boxes) Highlight the five enhancers E1–E5 defined by these histone signatures.
Nucleosomes harboring histone H3 lysine 4 monomethylation (H3K4me1) define enhancers (The ENCODE Project Consortium 2007; Heintzman et al. 2007, 2009; Lupien et al. 2008). Furthermore, this epigenetic modification is distributed across the genome in a tissue-type-specific manner, guiding tissue-type-specific transcriptional programs (Lupien et al. 2008; Heintzman et al. 2009; Ernst et al. 2011). To determine if the rs1859962 PCa risk LD block harbors enhancers, we examined the H3K4me1 genome-wide distribution defined by ChIP-seq assays in LNCaP and VCaP PCa cells (Yu et al. 2010). We also included in our analysis the genome-wide H3K4me1 distribution from 10 additional cell lines of different origin (H1-hESC, HMEC, HSMM, HSMMtube, HUVEC, K562, NH-A, NHEK, NHLF, and Osteoblast) (The ENCODE Project Consortium 2007). We show that there are five enhancers within the target genomic segment, hereafter referred to as E1–E5 (Fig. 1D; Supplemental Fig. S4). Comparisons between PCa cells and the 10 additional cell lines demonstrate that the E1 and E2 enhancers are specific to PCa cells, while E3–E5 are exclusively found in non-PCa cells (Fig. 1D; Supplemental Fig. S4). In addition to marking silenced genes, H3K9me3 can also associate with silenced enhancers (Zentner et al. 2011). The E1 and E2 enhancers lack enrichment with H3K9me3 in PCa cells, suggesting that they are active regulatory elements within these cells (Fig. 1C). Regulatory elements such as enhancers permissive to transcription factor binding are commonly found in regions of open chromatin, which can be identified by genome-wide DNase I hypersensitivity assays combined to massively parallel sequencing (DNase I-seq) (The ENCODE Project Consortium 2007; Hesselberth et al. 2009; Boyle et al. 2011; Pique-Regi et al. 2011). The ENCODE project recently released the DNase I-seq data from 77 human cell lines generated from diverse tissues, including the LNCaP PCa cells (The ENCODE Project Consortium 2007). Focusing on the rs1859962 PCa risk LD block, DNase I-seq identifies the E1–E5 enhancers and confirms that the E1 and E2 enhancers are present in PCa cells (Supplemental Fig. S5). Incidentally, rs1859962 risk variant maps directly to the E1 enhancer (Fig. 1). Overall, while multiple enhancers are found within the rs1859962 PCa risk LD block, only E1 and E2 are present in PCa cells.
The E1 enhancer forms a long-range chromatin loop to the SOX9 gene
“Enhancers” are defined as regulatory elements that can potentiate expression of genes regardless of distance or orientation (Blackwood and Kadonaga 1998; Bulger and Groudine 1999; Heintzman et al. 2007). Distant enhancers can work through long-range chromatin loops to physically interact with promoters of target genes (Dekker et al. 2002; Kagey et al. 2010). These can be detected using Chromatin Conformation Capture–based assays (3C). Briefly, through cross-linking, digestion, and ligation, DNA fragments that are frequently in spatial proximity of each other are more likely to be ligated together than DNA fragments that are not (Dekker et al. 2002). These ligated fragments can then be detected by PCR and/or sequencing-based approaches. Using a TaqMan probe–based 3C-qPCR assay in LNCaP cells, we investigated all possible chromatin loops between the E1 enhancer and surrounding genes within an ∼3-Mb window. Of all the pairwise comparisons, we detected a single chromatin loop spanning 1 Mb between E1 and the promoter of the SOX9 gene (Fig. 2A,B; Supplemental Fig. S8). This chromatin loop was confirmed through TaqMan probe–based qPCR assays with a probe targeting the SOX9 gene (Fig. 2C). The chromatin interaction was further confirmed by sequencing the PCR product (Supplemental Fig. S7). Furthermore, this chromatin loop appears to be specific to SOX9, because no other loop could be detected between E1 and any other genes (MAP2K6, KCNJ16, KCNJ2) found within this ∼3-Mb window (Fig. 2A,B). This parallels the fact that SOX9 is the only gene in this ∼3-Mb window that is overexpressed in primary prostate tumors compared with normal prostate epithelium (Fig. 2A; Yu et al. 2004; Taylor et al. 2010). In addition, none of the other enhancers (E2–E5) were involved in long-range chromatin loops with the SOX9 gene (Fig. 2C). Overall, these results suggest that the rs1859962 PCa risk LD block targets SOX9 through a long-range chromatin loop connecting it to the E1 enhancer.
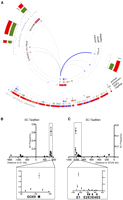
The E1 enhancer is connected to the SOX9 gene through a 1-Mb chromatin loop. (A) Circular representation of the 3-Mb DNA region surrounding the 17q24.3 risk locus. From inner to outside: chromatin loops identified by TaqMan probe–based 3C-qPCR in LNCaP cells (lines correspond to tested loops). (Blue) A detected loop; (gray) undetected loops. Locations of 3C primers, genes and enhancers, the LD block defined by the International HapMap Project for the rs1859962, and population expression level of nearby genes; (green) normal prostate epithelium; (red) primary prostate tumors. For gene expression, the y-axis represents the individual log2 expression level ±1 SEM. Expression levels from prostate tumors are compared with normal samples. The P-value is derived from a t-test; (***) p ≤ 0.001. Note the negative level of MAP2K6, KCNJ16, and KCNJ2 expression. (Bottom panel) Zooms into the LD block associated with rs1859962 (red bar) defining the SNPs in LD (black bars). LD score: (dark red) LOD = 2, D′ = 1; (light red) LOD = 2, D′ < 1; (blue) LOD < 2, D′ = 1; (white) LOD < 2, D′ < 1. (B) 3C-TaqMan assays with a TaqMan probe targeting the E1 enhancer. (X-axis) Genomic distance from each 3C test site to the E1 enhancer. (Y-axis) 3C interaction frequency. The SOX9 gene region is highlighted in the bottom panel. (C) Same as B but with regard to a TaqMan probe targeting the SOX9 gene. The 17q24.3 PCa risk LD block is highlighted in the bottom panel.
Two SNPs within the E1 enhancer impose allele-specific gene expression
Based on the striking relationship uncovered between E1 and SOX9, we next sought to determine if any potential causative SNP(s) in the vicinity of E1 could be driving the risk associated with the 17q24.3 risk LD block in PCa. Previous reports provided evidence that disruption of transcription factor recruitment may be associated with cancer risk variants (Rahimov et al. 2008; Jia et al. 2009; Pomerantz et al. 2009; Tuupanen et al. 2009; Wright et al. 2010; Harismendy et al. 2011). Since transcription factors normally bind to open chromatin, we first identified the specific regions of DNase I hypersensitivity (DHS) within the E1 enhancer. This analysis revealed two distinct regions: one of 360 bp (Chr17: 69,107,686–69,108,045) and another of 300 bp (Chr17: 69,108,466–69,108,765) (Fig. 3A). Luciferase reporter constructs containing either of these regions demonstrate that both have enhancer activity when transfected in LNCaP PCa cells (Fig. 3B). Taking into account all known SNPs (dbSNP132) (Sherry et al. 2001), we found five SNPs mapping to these DHS regions (Fig. 3A). To identify which of these could disrupt gene expression, we performed site-directed mutagenesis to introduce the variant allele of each of these five SNPs in our luciferase reporter constructs. Through luciferase assays in LNCaP cells transfected with either the reference or the variant reporter constructs, we detected allele-specific changes in expression for two of the five SNPs (Fig. 3B). Indeed, the variant alleles of both the rs8072254 and rs1859961 SNPs present in the 360-bp and 300-bp DHS, respectively, increased gene expression (Fig. 3B). This is also the case when luciferase plasmids containing the E1 enhancer and the SOX9 promoter were used (Supplemental Fig. S13), suggesting the enhancer activity of E1 for the SOX9 gene. These SNPs also reside at the center of their respective DHS regions and map to sites of evolutionary conservation across mammals (Fig. 3A). This further supports their biological relevance because evolutionary conservation is linked to biological function (Kolaczkowski and Kern 2010). Overall, this suggests that both variants have the potential to account for the risk associated with the rs1859962 SNP in PCa. Specifically, the variant allele of the rs8072254 and rs1859961 SNPs mapping to DHS sites within E1 can significantly disrupt enhancer activity to modulate target gene expression.
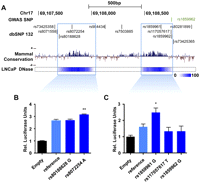
Candidate-functional variants within the E1 enhancer. (A) Overview of the E1 enhancer region. All SNPs from the dbSNP132 database are presented. Five are found in highly conserved regions across mammals (PhyloP value track presented) that map to the two DHS regions found in E1. The mammal basewise conservation score is presented according to their PhyloP value. (B,C) Luciferase assays using plasmids harboring the two DHS regions and either the reference or variant sequence for the five SNPs found within them. The pGL3 plasmid without the enhancer region (Empty) is used as a negative control. (Y-axis) Relative luciferase units normalized to Renilla signal ±1 SEM. The luciferase expression level for each variant SNP is compared with the plasmid homozygous for the reference alleles. The P-value is derived from a t-test; (*) p ≤ 0.05; (**) p ≤ 0.01.
Two candidate causal variants alter transcription factor recruitment
Transcription factors bind to chromatin by recognizing specific DNA sequences known as DNA motifs (Vaquerizas et al. 2009). Transcription factor recruitment can be disrupted by SNPs mapping to these DNA motifs (Rahimov et al. 2008; Jia et al. 2009; Cowper-Sal·lari et al. 2010; Wright et al. 2010; Freedman et al. 2011). The probability that a given SNP will modify transcription factor binding may be inferred using the position-weighted matrix (PWM) for any given DNA motif (Kasowski et al. 2010). PWM represents the frequency distribution of nucleotides at each position of transcription factor binding sites (Stormo and Fields 1998).
The rs8072254 SNP maps to the third position of the androgen response element (ARE) DNA motif recognized by the androgen receptor (AR), an essential transcription factor in the initiation and progression of prostate cancer (Fig. 4A; Heinlein and Chang 2004; Wang et al. 2009). The ARE PWM, defined ChIP-seq assays against AR, reveals that the variant adenine (A) allele of rs8072254 is favored compared with its reference guanine (G) allele by AR (Fig. 4A). In vitro–directed allele-specific ChIP assays using the luciferase reported constructs for the rs8072254 SNP demonstrates that while both the reference G and variant A allele of rs8072254 are permissive for receptor binding, AR binds to the variant A allele with a significantly higher affinity than the reference G allele (Fig. 4B). In agreement, the luciferase assay in LNCaP cells depleted or not of AR using siRNA reveals that AR is required for the increased transcriptional response associated with the rs8072254 variant A allele (Fig. 4C). This suggests that increased AR binding to the E1 enhancer due to the rs8072254 variant A allele can drive an increased expression of the target gene.
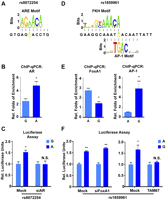
The functional SNPs modulate transcription factor bindings, further imposing allele-specific gene expression. (A) The PWM matrixes of ARE DNA motif overlapping with rs8072254 are indicated. The alleles of rs8072254 are presented; (yellow) reference G allele; (green) variant A allele. (B) In vitro allele-specific AR ChIP-qPCR assay of the two alleles from rs8072254. DNA enrichment is normalized to the DNA input. (Y-axis) Relative folds of enrichment with regard to FOXA1 ± 1 SEM. The P-value is derived from a t-test; (*) p ≤ 0.05. (C) A luciferase assay was performed in PCa LNCaP cells. The experiments were transfected with control (Mock) and AR siRNA, respectively. Luciferase units were normalized to Renilla signal and further normalized to the G allele of rs8072254 ± 1 SEM. The P-value is derived from a t-test; (*) p ≤ 0.05; (N.S.) non-significant. (D) The PWM matrixes of FKH and AP-1 DNA motif overlapping with rs1859961 are indicated. The alleles of rs1859961 are presented; (green) reference A allele; (yellow) variant G allele. (E) Same as B but with regard to FOXA1 and AP-1 ChIP-qPCR assay of the two alleles from rs1859961; (*) p ≤ 0.05. (F) A luciferase assay was performed in PCa LNCaP cells. (Left panel) The experiments were transfected with Control (Mock) and FOXA1 siRNA, respectively. (Right panel) The experiments were treated with an empty control plasmid and the dominant-negative AP-1 mutant TAM67 plasmid, respectively. Luciferase units were normalized to Renilla signal, and further normalized to the A allele of rs1859961 ± 1 SEM. The P-value is derived from a t-test; (*) p ≤ 0.05; (**) p ≤ 0.01; (N.S.) non-significant.
rs1859961 maps to the tenth position of the forkhead (FKH) DNA motif (Fig. 4D). The forkhead family member FOXA1 plays an essential role in PCa cells, acting as a pioneer factor by opening the chromatin and positioning nucleosomes to favor transcription factor recruitment to promote growth of PCa cells (Lupien et al. 2008; Wang et al. 2009; He et al. 2010). The FKH PWM, defined based on thousands of FOXA1 binding sites identified by ChIP-seq assays, reveals that the tenth position is invariably an A residue (Fig. 4D). The rs1859961 variant allele being a G suggests that it should significantly impair FOXA1 binding. In vitro–directed allele-specific ChIP assays using the luciferase reported constructs for the rs1859961 SNP demonstrate that FOXA1 preferentially binds to the reference A versus variant G allele (Fig. 4E; Supplemental Fig. S14). Hence, reduced FOXA1 binding to the chromatin is associated with higher transcriptional activity for the E1 enhancer. Although FOXA1 can act as a repressor (Malik et al. 2010; Lupien et al. 2008; Magnani et al. 2011), this is in sharp contrast with its activator role characterized to date in PCa cells. To test FOXA1's capacity to act as a repressor, we measured the luciferase activity from the rs1859961 SNP in LNCaP cells depleted or not of FOXA1 using control and FOXA1 siRNAs, respectively. FOXA1 depletion did not alter the allele-specific expression driven by the rs1859961 SNP (Fig. 4F). This suggests that the allele-specific binding of FOXA1 within the E1 enhancer does not account for the allele-specific expression driven by the rs1859961 SNP.
The DNA motif overlap analysis also reveals that rs1859961 maps to an AP-1 DNA motif. As a result, the variant G allele effectively creates an AP-1 DNA motif (Fig. 4D). The AP-1 transcription factor, composed of c-JUN, c-FOS and ATF subunits, is associated with PCa progression (Ouyang et al. 2008). To demonstrate that rs1859961 can modulate AP-1 chromatin binding, we performed in vitro allele-specific directed ChIP assays. These reveal the preferential recruitment of AP-1 to the variant G allele compared with the reference A allele (Fig. 4E). The luciferase reporter assay performed in LNCaP cells transfected or not with a dominant-negative form of AP-1 (TAM67) (Dhar et al. 2004) shows that AP-1 is driving the increased transcriptional response associated with the rs1859961 variant allele (Fig. 4F). Indeed, the increased luciferase expression associated with the variant G allele is brought back to the same level as for the reference A allele in LNCaP cells ectopically expressing the dominant-negative AP-1 (TAM67) (Fig. 4F). This suggests that increased AP-1 binding to the E1 enhancer due to the rs1859961 variant G allele can drive an increased expression of the target gene. Overall our results suggest that the rs8072254 and rs1859961 SNPs are functional within the 17q24.3 risk LD block because they modulate AR and AP-1 binding, respectively, and this leads to an increased transcriptional activity of the E1 enhancer that loops to the SOX9 oncogene.
Using the 1000 Genomes Project data sets (The 1000 Genomes Project Consortium 2010), we extracted the genotype data for the 17q24.3 region to determine the LD between both the rs1859962 risk SNP and either the rs8072254 or rs1859961 SNPs. The variant A allele of rs8072254 is common (minor allele frequency [MAF] = 0.36–0.5) and in strong LD (r2 ≥ 0.96) with the risk G allele of the rs1859962 PCa risk SNP among individuals of European ancestry (Supplemental Fig. S9; Supplemental Table S1), supporting its relevance to the observed PCa risk called in populations of European ancestry. In fact, a perfect association (r2 = 1) between the functional A allele of rs8072254 and the risk G allele of rs1859962 is detected in all populations of European ancestry (British from England and Scotland [GBR], Iberian populations in Spain [IBS], Toscani in Italia [TSI], and Utah residents with Northern and Western European ancestry [CEU]), except the Finnish from Finland (FIN) (Supplemental Table S1). However, the functional G allele of rs1859961 is rare (MAF ≤ 0.012) and was only present in Colombians (Colombians in Medellin, Colombia [CLM]) and several groups with African ancestry (African Ancestry in Southwest United States [ASW], Luhya Webuye Kenya [LWK], Yoruba in Ibadan, Nigeria [YRI]). Furthermore the G allele of rs1859961 is only associated with the reference T allele of rs1859962 (D′ = 1, r2 = 0.004–0.009) (Supplemental Table S1). Overall this suggests that the rs8072254 SNP can account for the risk associated with the rs1859962 PCa SNP in individuals of European ancestry by increasing enhancer activity, while the rs1859961 SNP can also influence enhancer activity but can only disrupt the function of this LD block in populations of African and Colombian ancestry.
Discussion
While genome-wide association studies define genetic risk variants that account for the heritability of diseases, they rarely identify the targeted functional biology because most risk variants map to poorly annotated noncoding genomic regions (Hindorff et al. 2009). Using an integrative functional genomics approach that combines epigenomic, open chromatin, and haplotype mapping with GWAS results, we show that the risk associated with the PCa rs1859962 risk LD block is in part accounted for by multiple genetic variants mapping to a unique enhancer looping to the SOX9 oncogene. Combined with other recent findings that identified cis-regulatory elements in 8q24.3 PCa- and 9p21 coronary artery disease–risk loci (Jia et al. 2009; Pomerantz et al. 2009; Tuupanen et al. 2009; Wright et al. 2010; Harismendy et al. 2011), our study demonstrates that targeting distant regulatory elements, such as enhancers, is a common mechanism of action of genetic risk variants. Importantly, two functional variants (rs8072254 and rs1859961) can increase the enhancer activity, suggesting that both have the potential to contribute to the risk of PCa associated with this locus (Fig. 5).
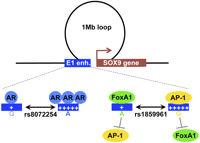
Model for the functional SNPs rs8072254 and rs1859961. The E1 enhancer within the 17q24.3 risk locus loops to the SOX9 gene. Within the E1 enhancer, the variant A allele of rs8072254 increases the binding of AR and further drives the increased enhancer activity. The variant G allele of rs1859961 decreases the binding of FOXA1 but increases the binding of AP-1. This latter increases enhancer activity. This E1 enhancer establishes a loop with the SOX9 genes in prostate cancer cells.
Risk variants mapping to noncoding regions may also directly impact unannotated transcripts such as lincRNAs or miRNAs (Cabili et al. 2011; Freedman et al. 2011). For instance, genetic variants in miRNAs have been shown to decrease miRNA expression and to be associated with a higher risk of papillary thyroid carcinoma (Jazdzewski et al. 2008). While we could identify two SNPs disrupting enhancer activity in the rs1859962 risk LD block, we could not identify any sign of transcription in PCa cells or any other cell type based on epigenomic signatures within this region.
Our study revealed that the PCa rs1859962 risk LD block loops to the SOX9 gene, specifically through the E1 enhancer. Previous studies have shown that SOX9 is highly relevant to PCa development. Its overexpression in PCa cells promotes tumor growth and invasion, while its depletion hinders prostate tumor growth (Wang et al. 2007, 2008; Thomsen et al. 2010). At the protein level, it contributes to AR expression, central to prostate cancer development (Wang et al. 2007). SOX9 can also cooperate with the loss of the tumor suppressor PTEN to further drive prostate tumor formation (Thomsen et al. 2010). Furthermore, the SOX9 gene is significantly overexpressed in primary prostate tumors compared with normal prostate epithelium (Yu et al. 2004). Moreover, overexpression of SOX9 in combination with SLUG promotes the reprogramming of breast epithelial cells, suggesting SOX9's role in developing cancer stem cells (Guo et al. 2012). Together, our results suggest that the risk variants disrupting E1 enhancer activity can impact the expression of target transcripts such as the SOX9 gene. Future efforts through improved genome-wide technologies characterizing changes in expression profile based on the genetic identity of PCa patients, such as e-QTL analysis (expression quantitative trait loci) or unbiased genome-wide chromatin conformation capture assays, may likely identify additional targets of this enhancer.
Understanding cis-regulatory mechanisms underlying risk variants mapping to functional regulatory elements is key in characterizing their causality. Previous studies have demonstrated that functional SNPs can either favor or repress transcription factor binding with direct consequence on enhancer activity (Rahimov et al. 2008; Jia et al. 2009; Pomerantz et al. 2009; Tuupanen et al. 2009; Wright et al. 2010; Harismendy et al. 2011) such as we describe for the rs8072254 SNP (Fig. 5). Previous studies have suggested that the gained AR binding in the SOX9 gene body after FOXA1 silencing up-regulates the SOX9 expression (Wang et al. 2011). Our results suggest that the AR binding gained by a functional SNP (rs8072254) 1 Mb away from the SOX9 gene may also contribute to the up-regulation of the SOX9 expression. In addition, our results revealed another layer of complexity in enhancer activity associated with functional SNPs. In the case of rs1859961, the variant allele can both repress the binding of FOXA1 while favoring the binding of the AP-1 transcription factor (Fig. 5). This can be predicted purely based on sequence analysis and DNA motif PWM scores. This suggests that characterization of the biology disrupted by risk variants would benefit from the generation of a map of disruptive SNPs based on their capacity to alter PWM scores for each known DNA motif.
Overall, we identified the cis-regulatory features underlying the 17q24.3 PCa risk locus, further supporting the integrative functional genomics approach in post-GWAS studies. In addition, this methodology is compatible with the identification of the functional biology disrupted by mutations. This approach will need to be integrated within the analysis pipeline from tumor whole-genome resequencing efforts led by the ICGC (International Cancer Genome Consortium), TCGA (The Cancer Genome Atlas), and COSMIC (Catalogue of Somatic Mutations in Cancer) projects that are cataloguing somatic mutations including coding and noncoding mutations (Hudson et al. 2010; The Cancer Genome Atlas Research Network 2011; Forbes et al. 2011).
Methods
Cell culture and transfection
LNCaP cells were cultured in RPMI media with 10% FBS supplemented. For plasmid transfection, LNCaP cells were transfected with the mock (pcDNA3.1) or AP-1 dominant-negative (TAM67) vectors (1 μg per well for six-well plates) using LipoD293 DNA transfection reagent according to the manufacturer's recommendations (SignaGen). Luciferase assays were performed 48 h after transfection. For small-interfering RNA (siRNA) transfection, LNCaP cells were transfected with scrambled siRNA (siControl) or siFOXA1 (final concentration: 24 nM) using the Lipofectamine 2000 Kit (Invitrogen). RNA was extracted 48 h after transfection. Previously validated RNA oligonucleotides against FOXA1 were used (Lupien et al. 2008): sense: 5′-GGACUUCAAGGCAUACGAAUU-3′; antisense: 5′- UUCGUAUGCCUUGAAGUCCUU-3′.
Calling SNPs in linkage disequilibrium: LDX tool
Linkage Disequilibrium eXtend (LDX) is a command-line tool we created to query the UCSC Genome Browser's CEPH HapMap Linkage Disequilibrium Phase II table (Hg19, hapmapLdPhCeu). Given a SNP identifier, the program returns the list of SNPs that satisfy LOD > 2 and D′ > 0.99 criteria.
LD analysis from the 1000 Genomes Project
The current release (February 2012) of the genotype calls from the 1000 Genomes Project was downloaded from the project's website (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/). LD was calculated using phased haplotypes with vcftools (Danecek et al. 2011), only unrelated individuals were included in the estimation of the allele frequencies and the LD calculations. Individuals identified by Pemberton et al. (2010) to be related in the subset of individuals shared between the current release and HapMap phase III were removed.
H3K36me3 ChIP-seq assays
ChIP-seq assays were performed as previously described (He et al. 2010). Briefly, LNCaP cells were treated with MNase (Sigma-Aldrich N3755), and ChIP was performed with H3K36me3 antibody (Abcam Ab9050). Libraries were prepared for the Illumina Genome Analyzer according to the manufacturer's instructions. Sequencing reads were aligned to the human genome (Hg19) using the Illumina Genome Analyzer Pipeline. Regions significantly enriched (p = 1 × 10−5) for H3K36me3 were detected using the MACS software (Zhang et al. 2008).
Epigenomics and chromatin openness data
LNCaP and VCaP (Korenchuk et al. 2001) ChIP-seq data were extracted from the online genomic resource Nuclear Receptor Cistrome (http://cistrome.dfci.harvard.edu/NR_Cistrome). The ChIP-seq and DNase I-seq data for all cell lines were extracted from the ENCODE Project (http://genome.ucsc.edu/ENCODE) (The ENCODE Project Consortium 2007). The ENCODE Project data were displayed on the UCSC Genome Browser (http://genome.ucsc.edu). LNCaP and VCaP ChIP-seq data were displayed on the IGV Browser (http://www.broadinstitute.org/igv).
Gene expression analysis in prostate tumor samples
Gene expression data were extracted from the online resource ONCOMINE (https://www.oncomine.com/resource). The expression data of MAP2K6, KCNJ2, and SOX9 were extracted from Yu's prostate cancer study (Yu et al. 2004), which includes 23 normal prostate glands and 65 primary prostate tumors and covers 8603 genes using the Human Genome U95A-Av2 array. The expression data of KCNJ16 were extracted from Taylor's prostate cancer study (Taylor et al. 2010), which includes 29 normal prostate glands and 131 prostate tumors and covers 22,238 genes using the Affymetirix Human Exon 1.0 ST array.
3C-qPCR assays
Chromosome conformation capture (3C) technology coupled to qPCR was performed according to a published protocol (Hagege et al. 2007). Briefly, 5 million LNCaP cells were processed using formaldehyde cross-linking (1%, 10 min at room temperature) of interacting chromatin segments, followed by HindIII digestion (400 units, overnight at 37°C) and ligation (T4 DNA ligase 4000 units, 4 h at 16°C). DNA fragments were cleaned by phenol:chloroform, followed by ethanol extraction. TaqMan probe–based qPCR was performed to quantify the ligated DNA fragments. Briefly, the TaqMan probe targets the strand antisense to the PCR primer, but in the same restriction fragment. Thus the fluorescent signal released during the PCR elongation step is specific to the amplicons of the relevant ligation fragments. Probes used in the TaqMan assays are listed in Supplemental Table S2. The qPCR result from the 3C-processed sample was normalized to BAC templates according to previously published literature (Dekker et al. 2002; Kagey et al. 2010). The ligation products from digested BAC clones (RP11-660P13, RP11-939115, RP11-134J16, RP11-295K15, and RP11-300G13) were diluted to the same molar concentration as the 3C sample before qPCR. Then, in order to control the qPCR efficiency, we normalized the ligation frequency by dividing the qPCR signal from the 3C sample (triplicates) by that from the BAC controls. DNA gel after PCR amplification of BAC templates confirmed that all PCR amplicons using our PCR primers have correct sizes (Supplemental Figs. S10, S11). The primers for 3C-qPCR are listed in Supplemental Table S2.
To confirm the observed E1-SOX9 chromatin interaction, we used another normalization method for 3C data analysis (Hagege et al. 2007). Briefly, we generated the Ct value standard curve using BAC libraries for each tested ligation. Then we quantified the ligation products between the E1 enhancer and each of the tested 3C sites using the parameters from each corresponding standard curve (b: intercept, a: slope): value = 10(Ct − b)/a. These values are further normalized toward an internal control. (We used primers hybridized to the ACTIN gene [Supplemental Fig. S8].)
Luciferase assay
Two fragments covering the left and right DNase I–hypersensitive sites within the E1 enhancer were PCR-amplified from LNCaP cells and cloned into the minimal promoter containing firefly Luciferase reporter vector pGL3 (Promega). These regions are homozygous for the major alleles of each target SNP. Site-directed mutagenesis (Agilent Technologies QuickChange Kit) was performed to generate the plasmids homozygous for the minor allele of each target SNP: rs80188628 T>G; rs8072254 G>A; rs1859961 A>G; rs117057617 C>T; and rs1859962 T>G (Supplemental Fig. S12). Each construct was cotransfected into PCa LNCaP cells with the Renilla reporter construct to perform luciferase assays according to the manufacturer's recommendations (Promega). Primers used in cloning and mutagenesis are listed in Supplemental Table S2.
Allele-specific ChIP-qPCR
FOXA1 (Abcam, ab5089) and AP-1 (Santa Cruz, sc44 and sc253) ChIP-qPCR was performed as previously described (Lupien et al. 2008, 2010). An allele-specific MAMA PCR-based technique was applied to assess differential FOXA1 enrichment on heterozygous alleles (Li et al. 2004). The forward primers are allele-specific MAMA primers that were designed as previously described (Li et al. 2004). The reverse primer is complementary to the sequence from the plasmid. The primers are listed in Supplemental Table S2.
Data access
The ChIP-seq data sets we generated against H3K36me3 in LNCaP cells can be accessed through the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE35829.
Acknowledgments
We thank Dr. Jerome Eeckhoute (INSERM, Université de Lille), Dr. Mathieu Lemaire (Yale University), and Dr. Matthew Freedman (Dana-Farber Cancer Institute) for discussions; and Dr. Nancy Colburn (NCI-Frederick) for providing the dominant-negative AP-1 construct (TAM67). This work was supported by grants from the NCI (R01CA155004 to M.L.), the ACS (IRG-82-003-27 to M.L.), and the NLM (R01LM009012 to J.H.M.).
Footnotes
-
↵5 Corresponding author
E-mail mlupien{at}uhnres.utoronto.ca
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.135665.111.
- Received December 1, 2011.
- Accepted May 22, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








