
A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver
- Donatella Canella1,5,
- David Bernasconi1,2,5,
- Federica Gilardi1,6,
- Gwendal LeMartelot3,6,
- Eugenia Migliavacca2,6,
- Viviane Praz1,6,
- Pascal Cousin1,
- Mauro Delorenzi2,4,8,
- Nouria Hernandez1,8 and
- The CycliX Consortium7
- 9Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, 1015 Lausanne, Switzerland.
- 10Swiss Institute of Bioinformatics, University of Lausanne, Lausanne, Switzerland.
- 11Interfaculty Institute of Bioengineering, School of Life Sciences, Ecole polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
- 12Vital IT, Swiss Institute of Bioinformatics, Lausanne, Switzerland.
- 13Bioinformatics and Biostatistics Core Facility, School of Life Sciences, Ecole polytechnique Fédérale de Lausanne, Lausanne, Switzerland.
- 14Department of Molecular Biology, Faculty of Sciences, University of Geneva, Lausanne, Switzerland.
- 15Département de formation et de recherche, Centre Hospitalier Universitaire Vaudois and University of Lausanne, Lausanne, Switzerland
- 1Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, 1015 Lausanne, Switzerland;
- 2Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland;
- 3Department of Molecular Biology, Faculty of Sciences, University of Geneva, 1211 Geneva, Switzerland;
- 4Département de formation et de recherche, Centre Hospitalier Universitaire Vaudois and University of Lausanne, 1011 Lausanne, Switzerland
Abstract
The genomic loci occupied by RNA polymerase (RNAP) III have been characterized in human culture cells by genome-wide chromatin immunoprecipitations, followed by deep sequencing (ChIP-seq). These studies have shown that only ∼40% of the annotated 622 human tRNA genes and pseudogenes are occupied by RNAP-III, and that these genes are often in open chromatin regions rich in active RNAP-II transcription units. We have used ChIP-seq to characterize RNAP-III-occupied loci in a differentiated tissue, the mouse liver. Our studies define the mouse liver RNAP-III-occupied loci including a conserved mammalian interspersed repeat (MIR) as a potential regulator of an RNAP-III subunit-encoding gene. They reveal that synteny relationships can be established between a number of human and mouse RNAP-III genes, and that the expression levels of these genes are significantly linked. They establish that variations within the A and B promoter boxes, as well as the strength of the terminator sequence, can strongly affect RNAP-III occupancy of tRNA genes. They reveal correlations with various genomic features that explain the observed variation of 81% of tRNA scores. In mouse liver, loci represented in the NCBI37/mm9 genome assembly that are clearly occupied by RNAP-III comprise 50 Rn5s (5S RNA) genes, 14 known non-tRNA RNAP-III genes, nine Rn4.5s (4.5S RNA) genes, and 29 SINEs. Moreover, out of the 433 annotated tRNA genes, half are occupied by RNAP-III. Transfer RNA gene expression levels reflect both an underlying genomic organization conserved in dividing human culture cells and resting mouse liver cells, and the particular promoter and terminator strengths of individual genes.
RNA polymerase III (RNAP-III) synthesizes short RNAs involved in essential cellular processes, including protein synthesis, RNA maturation, and transcriptional control, but until recently, the full extent of the genomic loci occupied, and therefore probably transcribed, by RNAP-III in vivo, was unknown. Several groups have now used the ChIP-seq technique, i.e., chromatin immunoprecipitation followed by deep sequencing, to localize genome-wide RNAP-III and some of its transcription factors in several human cultured cell lines. These experiments have revealed a relatively modest number of new RNAP-III transcription units, from a few 10s to about 200, depending on the criteria applied (Barski et al. 2010; Canella et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010). In addition, most previously known RNAP-III genes were occupied by RNAP-III. Thus, in such cells RNAP-III and some of its transcription factors occupied 17 RN5S loci annotated on chromosome 1 in the NCBI/hg18 genome assembly, the VTRNA1-1, VTRNA1-2, VTRNA1-3, and VTRNA2-1 (hsa-mir-886) genes coding for vault RNAs, three SRP genes, 14 SNAR genes, five RNU6 genes, the RNU6ATAC gene, the RN7SK, RMRP, and RPPH1 genes, and the RNY1, RNY3, RNY4, and RNY5 (hsa-mir-1975) genes. Noticeably, however, a large fraction of the annotated tRNA genes was devoid of RNAP-III (Barski et al. 2010; Canella et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010), a phenomenon that seemed not easily explained by different qualities of the promoters, as the A and B boxes that constitute tRNA gene promoters were nearly identical in occupied and nonoccupied genes (Canella et al. 2010; Oler et al. 2010). Instead, RNAP-III-occupied tRNA genes were found to differ from non—or poorly occupied—genes by their proximity to peaks of RNAP-II occupancy and by their location in chromatin regions rich in histone marks typical of active RNAP-II promoters and enhancers (Barski et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010). This revealed that active RNAP-III genes are in chromatin regions that are very similar to those of active RNAP-II genes, often close to active RNAP-II transcription units, and led to the suggestion that active chromatin, probably established by RNAP-II transcription units, gates the access of RNAP-III to the genome (Oler et al. 2010).
The model above leaves several questions unanswered. Firstly, the previous experiments were performed in cultured cell lines. An important question is whether the set of RNAP-III-occupied loci is similar in a differentiated tissue. Secondly, it is still unclear why, in some cases, tRNA genes very close to each other, and thus in a similar chromatin environment, can be very differently occupied by RNAP-III. To address these issues, we have localized, genome wide, two subunits of RNAP-III, a subunit of RNAP-II, and the histone marks H3K4me3 and H3K36me3 in mouse liver cells. The results characterize RNAP-III-occupied loci in a normal mouse tissue. They reveal that synteny relationships can be established between a number of human and mouse RNAP-III genes, and that the expression levels of these genes are significantly linked. They also point to an RNAP-III-specific MIR as a potential regulator of an RNAP-III subunit-encoding gene. They reveal correlations between RNAP-III occupancy and a number of features, which, when analyzed in a multivariable regression model, account for ∼81% of observed variation of the tRNA gene scores. The results suggest that both an underlying genome organization, which is quite similar in human culture cells and in mouse liver cells, as well as features specific to individual genes including promoter and terminator strength, contribute to transcription efficiency of individual tRNA genes.
Results
To study genomic RNAP-III occupancy in mouse, we maintained C57/BL6 12–14-wk-old male mice as described in the Methods section, and collected livers from five mice at ZT02 (rep1) and, as a biological replicate, ZT26 (rep2). Formaldehyde cross-linked chromatin from each set of five livers was pooled and used as starting material for immunoprecipitations with antibodies directed against POLR3A (RPC1) and POLR3D (RPC4), two RNAP-III subunits, POLR2B (RPB2), the second largest RNAP-II subunit, as well as histone H3 trimethylated on lysine 4 (H3K4me3) or lysine 36 (H3K36me3). The starting decross-linked material, as well as the immunoprecipitated DNA, were then subjected to deep sequencing and analyzed as described previously (Canella et al. 2010; Supplemental Methods). The results allowed us to establish a list of RNAP-III-occupied loci in mouse liver consisting of tRNA genes (Supplemental Table S1), Rn5s genes (Supplemental Table S2), other known RNAP-III genes (Supplemental Table S3), Rn4.5s and related sequences (Supplemental Table S4), and B1, B2, and other SINE sequences (Supplemental Table S5), of which the most abundant were clearly tRNA genes (see Fig. 1A). Below, the terms “RNAP-III occupancy” or “RNAP-III transcription” are both used interchangeably. We cannot exclude, however, that in some cases the detected RNAP-III is paused, and thus not transcribing.
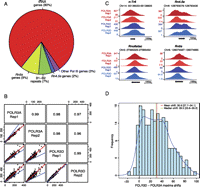
Identification of RNAP-III-occupied loci in mouse liver. (A) The pie chart summarizes the RNAP-III-occupied genomic regions in the mouse liver as determined by peak detection (Canella et al. 2010; see also Methods). The “other RNAP-III genes” category contains the known RNAP-III genes other than tRNA, Rn5s, and Rn4.5s (listed in Supplemental Table S3). (B) Pearson correlations of linear scores obtained with the anti-POLR3A and anti-POLR3D antibodies for the rep1 and rep2 biological replicates, as indicated. All loci listed in Supplemental Tables S1–S5 are included. The regression line is indicated in red, the y = x line in blue. The Pearson correlation coefficients are indicated in the squares in the top right. (C) UCSC browser views showing POLR3A and POLR3D peaks on a tRNA gene (n-Tr6, chr14, tRNA209-ArgACG), a Rnu6atac gene, a Rn4.5s gene, and a Rn5s gene. (D) The POLR3A and POLR3D peaks are offset relative to one another. The shift between the POLR3A and POLR3D peak summits (in a region from −30 to +70 around the TSS) for all tRNA genes with scores above 29.25 is shown on the x-axis, with the frequency on the y-axis. The mean and median shift values are indicated, together with the confidence interval (computed by the bootstrap method at 95% confidence interval).
We then calculated RNAP-III occupancy scores for the RNAP-III-occupied loci above as well as all 433 annotated tRNA genes, whether presenting an RNAP-III peak or not, as detailed in the Supplemental Methods. As shown in Figure 1B, we obtained correlations of 0.96 or higher between the linear scores obtained in the rep1 and rep2 experiments with the anti-POLR3A and anti-POLR3D antibodies for all the genes shown in Supplemental Tables S1–S5. The correlations for the log2 scores are shown in Supplemental Figure S1, and were 0.97 or higher. RNAP-III-occupied loci were divided into three tertiles, the lowest comprising loci with scores between 5 and 29.25, the second with scores between 29.25 and 115.36, and the highest with scores above 115.36.
Figure 1C shows a UCSC browser view of POLR3A and POLR3D peaks obtained on a tRNA (n-Tr6), a Rn4.5s, the Rnu6atac, and a Rn5s gene in the two biological replicates. Intriguingly, we observed that the POLR3A signal peaked slightly upstream of the POLR3D signal. To determine whether this was a general feature, we examined the shift between the POLR3A and POLR3D peak summits for all tRNA genes with scores above 29.25. As shown in Figure 1D, the mean shift between POLR3A and POLR3D peak maxima was 30.9 bp (confidence interval 27.7–34.1), with the median at 29.5 bp (confidence interval 23.8–35.2). This suggests that the complexes immunoprecipitated with anti-POLR3A antibodies are, on average, cross-linked to DNA upstream of complexes immunoprecipitated with anti-POLR3D antibodies, perhaps reflecting the distance between the two epitopes within the polymerase. For POLR3A and POLR3D peaks consisting mostly of unique tags, and whose shape is therefore reliable, we used the offset between the peaks as a criterion of bona fide RNAP-III occupancy.
RNAP-III-occupied loci in mouse liver: tRNA genes
The annotated mouse tRNA genes (http://gtrnadb.ucsc.edu/) comprise 430 standard tRNA genes, two selenocysteine tRNA genes and one possible suppressor tRNA gene, and are listed in Supplemental Table S1 together with the average scores for POLR3A and POLR3D for the biological replicates and the corresponding individual scores. Out of these genes, 177 had scores below five and were considered not transcribed (in pink in the first column), 48 belonged to the lowest tertile of all RNAP-III-occupied loci (orange in the first column), 100 to the second tertile (green in the first column), and 108 to the highest tertile (blue in the first column).
As previously observed in human culture cells (Canella et al. 2010), in all cases where there is a single gene corresponding to a certain isoacceptor (tRNAIle_GAT, tRNAVal_GAC, tRNAHis_ATG, and tRNASer_GGA, as well as tRNASup_TTA, marked in purple in column 2), this gene was not (or very poorly) transcribed. For all other isoacceptors, several corresponding genes were expressed, except for the tRNASeC_TCA, where only one gene (out of two, both marked in yellow in column 2) was expressed. The expressed tRNASeC_TCA gene has a type 3 promoter like its human counterparts, but the silent gene has, instead, internal A and B boxes. As expected, both initiator (marked in turquoise in column 1) and elongator tRNAMet genes were expressed.
RNAP-III-occupied loci in mouse liver: Rn5s genes
Out of the more than 1000 Rn5s and Rn5s-related sequences annotated in the mouse genome, only those that are part of a cluster of 50 annotated Rn5s genes on chromosome 8, between positions 126,062,686 and 126,147,645, displayed peaks (Supplemental Table S2). Among these 50 genes, the sequences within and surrounding the Rn5s coding sequences are highly similar, as shown in the alignment in Supplemental Figure S2. All genes display a “box D,” a sequence extending from −33 to −22 upstream of the transcription start site, conserved in the human, hamster, and mouse 5S genes, and required for efficient transcription of the human genes (Nielsen et al. 1993), as well as the internal promoter composed of Box A, the intermediate element IE, and box C (Bogenhagen et al. 1980; Sakonju et al. 1980; Pieler et al. 1987). However, 22 of these genes harbor mutations within the region extending from the D box to the end of the Rn5s-coding sequence.
To estimate transcription efficiency of these variant Rn5s sequences, we calculated the RNAP-III occupancy scores as above, as shown in Supplemental Table S2. Since in this case these scores are largely based on tags with multiples matches in the genome, they should be interpreted with caution. Nevertheless, some interesting patterns emerged. Thus, of the nine genes with the lowest scores, eight (Supplemental Table S2, genes highlighted in green in column 2) displayed mutations within box C not present in the other genes (see alignment in Supplemental Fig. S2). Moreover, the seven bottom ones (in blue in column 3) displayed additional mutations at or just downstream from the transcription start site (TSS). Thus, some of the Rn5s genes within the cluster appear less efficiently transcribed than others, most likely mainly as a result of mutations in box C, with perhaps an additional negative effect of mutations close to the TSS.
RNAP-III-occupied loci in mouse liver: Other known RNAP-III genes
Supplemental Table S3 lists the other known RNAP-III genes that displayed peaks. These comprise three genes with type 2 (in orange in the first column) and 11 genes with type 3 (in yellow in the first column), promoters, which, together with the tRNASeC_TCA on chromosome 7, brings the total of known RNAP-III genes with type 3 promoters clearly occupied by RNAP-III in mouse liver to 12. An alignment showing the sequences of the octamer, PSE, and TATA promoter elements in these genes is shown in Supplemental Figure S3. Supplemental Table S3 indicates the corresponding human syntenic genes, which could be clearly identified. As expected, the mouse BC1 RNA (Bc1) gene (chr7:100,808,784–100,808,949), which is specifically transcribed in neuronal cells in rodents (Anzai et al. 1986; Martignetti and Brosius 1995), did not display peaks. The genes in Supplemental Table S3 are briefly discussed in the order they appear in the table.
Human cells contain nine full-length U6 (RNU6) loci (Domitrovich and Kunkel 2003), of which we found five occupied by RNAP-III, three (RNU6-8, RNU6-9, and RNU6-2) at high levels (Canella et al. 2010). A BLAT search of the mouse genome for perfectly conserved Rnu6 RNA coding sequences revealed 51 loci. Of these, we again found five to be occupied by POLR3A and POLR3D, three with scores higher than 100. These five genes are syntenic with the five actively transcribed human RNU6 genes (see Supplemental Table S3). The gene with the second highest score corresponds to the previously studied mouse Rnu6 gene (Das et al. 1988). All five mouse Rnu6 genes have TATA boxes, PSEs, and octamer motifs, as shown in Supplemental Figure S3. However, in the two Rnu6 genes with the lowest scores, the spacing between the octamer and the PSE is unusuaI. The mouse Rnu6atac gene was highly occupied by POLR3A and POLR3D, suggesting high transcription rates (Supplemental Table S3, line 6), similar to what we observed previously for the syntenic human RNU6ATAC gene (Canella et al. 2010).
We found single genes occupied by POLR3A and POLR3D, corresponding to the previously described genes (Chang and Clayton 1989; Hsieh et al. 1990; Moon and Krause 1991) encoding each Mouse Rnu7sk (7SK) and Rmrp (RNase MRP) RNAs. Similarly, we found only one gene encoding Rpph1 (RNase P) occupied by POLR3A and POLR3D, corresponding to the previously described gene upstream of the Parp2 promoter and transcribed divergently (Altman et al. 1993; Ame et al. 2001). This gene is syntenic with the human RPPH1 gene, which is similarly transcribed divergently from the human PARP2 gene (see Supplemental Table S6 in Canella et al. 2010). Three genes reported to encode short Rpph1 RNA homologs (Li and Williams 1995) (accession numbers U31003, U31227, and U31228) did not display peaks and, indeed, have no convincing promoter sequences, suggesting that they are not true genes.
In human culture cells, four Y genes (RNY1–RNY4) are actively transcribed, of which RNY3 is the most highly transcribed, followed by RNY1 (Canella et al. 2010). In the mouse, two true Y RNA genes have been described, Rny1 and Rny3, as well as about 60 pseudogenes (Farris et al. 1996; Perreault et al. 2007). Only the Rny1 and Rny3 sequences, which are syntenic with human RNY1 and RNY3, were occupied by POLR3A and POLR3D, with Rny3 displaying the higher score (Supplemental Table S3, lines 10,11).
Mouse genes encoding Rn7sl (7SL) RNA have not been described, but the best genomic matches to human RN7SL RNAs (Ullu and Weiner 1985; Bredow et al. 1990; Englert et al. 2004) are two sequences on chromosome 12, each with the same four mismatches as compared with the human sequence. Both were occupied by POLR3A and POLR3D and are syntenic with the human RN7SL1 and RN7SL2 genes, which are by far the most highly transcribed of the three human RN7SL genes (Canella et al. 2010).
Among the four human vault genes (HVG-1–HVG-4), only HVG-1–HVG-3 were occupied by RNAP-III and its transcription factors (Canella et al. 2010). In mouse, two genes were identified, on chromosomes 8 and 18, of which only the second was expressed in mouse tissues (Kickhoefer et al. 2003). Consistent with these results, only the vault gene on chromosome 18 (Vaultrc5) was occupied by POLR3A and POLR3D (see Supplemental Table S3, row 14), and this gene is syntenic with the most highly expressed human vault gene, HVG-3. Thus, the 7SL, Y, and vault gene families contain fewer members in the mouse genome than in the human genome, and in each case the transcribed mouse members are syntenic with the most highly transcribed human members.
RNAP-III-occupied loci in mouse liver: Rn4.5s genes
Rn4.5s RNAs are present in four related rodent families, including the Muridae. They are synthesized by RNAP-III, but their function is unknown (Gogolevskaya and Kramerov 2010). In mouse, three true genes and a few hundred pseudogenes have been described (Gogolevskaya and Kramerov 2010, and references therein). The three described true genes are located on chromosome 6 and contain homologies to A and B boxes as well as two conserved elements, a GCC/AACGCCT and an AGAAT element located upstream of the TSS. These three Rn4.5s genes are annotated as B4A_SINE_B4 in the UCSC RNA table and are occupied by both POLR3A and POLR3D (see Supplemental Table S4, rows 1–3). In addition, we found six sequences on the same chromosome annotated 4.5SRNA_scRNA very clearly occupied by RNAP-III (rows 4–9), one of them 3′ of the previously described Rn4.5s genes, the others interspersed among them. These six Rn4.5s-related sequences are highly similar to each other but only weakly similar to the previously described Rn4.5s genes, as shown in the alignment in Supplemental Figure S4. They contain internal A and B boxes, but lack any obvious conserved motif with the 5′ flanking sequences.
RNAP-III-occupied loci in mouse liver: SINEs and other repeats
The mouse genome contains large numbers of short interspersed elements (SINEs) derived from RNAP-III transcription units by successive waves of retrotransposition. Those are classified into various families and classes. Repeats of the Alu family (B1 and B4 families in mouse) are derived from Rn7sl-encoding sequences, whereas repeats of the B2 family (B2 and B3 families in the mouse) are derived from tRNA sequences (Smit 1999). MIR family repeats are also derived from tRNA sequences and amplified before the eutherian radiation (Smit 1996). Among many sequences with peaks in the sissrs analysis, we selected those most convincingly occupied by RNAP-III (see Methods), which all have a distinct B box and often also a clear A box, as summarized in Supplemental Table S5. The few with high scores (rows 1–4) are all located within introns of RNAP-II transcription units. The most highly occupied SINE, the MIR listed in row 1, is intriguing. It is located on the minus strand within the first intron of the Polr3e gene, which encodes the POLR3E (RPC5) subunit of RNAP-III. As shown in Figure 2, A and B, human cells contain a MIR similarly located on the minus strand of the POLR3E gene (chr16:22,217,282–22,217440 in NCBI36/hg18), whose sequence can be aligned with that of the mouse MIR. We showed previously that this human MIR is occupied by RNAP-III as well as by BRF1 (see Supplemental Table S5, row 48 in Canella et al. 2010).
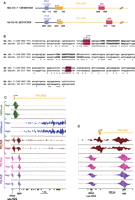
(A) Arrangement of RNAP-III genes in shared syntenic regions containing the Polr3e gene in the mouse and human genomes. The last four numbers of the chromosome coordinates are indicated below the lines. (B) Alignment of the mouse (Mus musculus, Mm) and human (Hs) MIR sequences. The region where RNAP-III transcription is likely to initiate and the terminator region are highlighted in red. Similarities to A- and B-box elements are indicated in bold. Identities are labeled with asterisks. (C) View of the tags obtained in the ChIPs performed with antibodies against the factors indicated on the left. The accumulations of + and − tags are shown above and below the lines, respectively. The location of the first exon and intron of the Polr3e gene is shown at top, that of the tRNA Leucine and the MIR at the bottom. (D) Zoom-in of the region around the Polr3e transcription start site.
To determine whether RNAP-III transcription of this MIR might affect RNAP-II transcription, we examined RNAP-II occupancy as well as the presence of H3K4me3 and H3K36me3 histone marks in this region. Figure 2, C and D shows tag accumulations over the beginning of the Polr3e gene. Consistent with active RNAP-II transcription, there is an increasing accumulation of the H3K36me3 mark in a 3′ direction within the body of the gene (Fig. 2C). There is also an accumulation of RNAP-II at the Polr3e promoter, as well as low RNAP-II occupancy over the body of the gene. There is, however, an unusual second accumulation of RNAP-II downstream of the promoter, which abuts precisely on the RNAP-III occupancy peak reflecting transcription of the MIR on the opposite strand (see Fig. 2D, which shows a zoom-in of the left part of Fig. 2C). Thus, active antisense RNAP-III transcription of the MIR apparently creates a barrier for RNAP-II, which, as a result, slows down and accumulates just at the downstream border of the MIR. Together with the observation that the MIR is conserved and occupied by RNAP-III in human cells, this suggests a role in Polr3e gene regulation.
Human–mouse shared syntenic RNAP-III genes have related RNAP-III scores
As shown above, we could easily define shared synteny relationships for known non-tRNA RNAP-III genes (see Supplemental Table S3). We examined whether this was also true for tRNA genes. We used the mouse–human chained alignments defined in the UCSC genome tables (Chiaromonte et al. 2002; Kent et al. 2003; Schwartz et al. 2003) and considered all tRNAs (Supplemental Table S1) and other known RNAP-III genes (Supplemental Table S3) in the mouse sequences. We then examined whether we could identify corresponding human genes in the shared syntenic regions. By considering only pairs of identical isoacceptors and positions relative to neighboring RNAP-II and other RNAP-III genes, we could assign clear shared synteny relationships for 229 tRNA genes. These are shown in Supplemental Table S6, which lists all chained alignments containing both mouse and human RNAP-III genes, with genes displaying shared synteny indicated in the last column.
Mouse tRNA genes are found on all chromosomes except chromosome 18, but the number of genes on the different chromosomes is highly variable. In particular, there are clusters of more than 10 tRNA genes on mouse chromosomes 1, 3, 6, 8, 11, 13, 19, and X. As shown in Supplemental Table S6, we could identify corresponding clusters, and in some cases corresponding genes in blocks of human–mouse shared syntenic regions. A striking example is that of the cluster on mouse chromosome 11, which, except for the first and last tRNA genes, is found in the reverse orientation on human chromosome 17 (Supplemental Table S6, see chain ID51). Similarly, the largest clusters of mouse tRNA genes, which are on chromosome 13, with 60 tRNA genes between positions 21,252,654 and 22,142,636 and 45 tRNA genes between positions 23,362,290 and 23,622,288, are found in large part in blocks of shared synteny on human chromosome 6, although, in this case, which exact mouse tRNA gene corresponds to which human tRNA gene is sometimes ambiguous (Supplemental Table S6, chain ID 98).
Casual inspection revealed similarities in RNAP-III occupancy of RNAP-III genes with shared synteny. For example, in the mouse, the 49 cysteines genes in the large cluster spanning 378,000 bp (from position 47,928,996 to 48,306,921) on chromosome 6 are silent except for the first one (chr6_157), which is well expressed (with a score of 113.77). The beginning of this cluster clearly corresponds with the beginning of a cysteine cluster on human chromosome 7 (see Supplemental Table S6, chain ID 14), where again only the first gene is significantly expressed (see Supplemental Table S3 in Canella et al. 2010). And, of the several tRNA genes located on chromosome X, only one was occupied by RNAP-III, the tRNA Val TAC gene on chrX; 157,204,042–157,204,114 in mouse and the same tRNA gene on chrX:18,602,950–18,603,022 in humans, which share synteny (see chain ID 29 in Supplemental Table S6). This prompted us to ask whether genes with shared synteny might have similar scores in human IMR90Tert cells and mouse liver cells. We used our previous data (Canella et al. 2010) and recalculated the scores for the human genes as described above for the mouse genes. We then eliminated those genes with >20% repeated tags, whose scores are, therefore, less reliable. We observed a moderate (Pearson's correlation coefficient = 0.49), but significant correlation (P-value 3.75 × 10−14, confidence interval 0.41–0.66). Thus, RNAP-III genes with shared synteny tend to display similar RNAP-III occupancy.
H3K4me3, but not H3K36me3, is present on RNAP-III-occupied isolated tRNA genes
The data above reveal that in the mouse liver, the RNAP-III-occupied loci comprise about 320 actively transcribed RNA RNAP-III genes. Strikingly, like in human cells where 43% of tRNA genes and pseudogenes are silent, a large fraction of mouse tRNA genes (41%) is not or little occupied by RNAP-III. We sought to understand why by examining possible correlations between RNAP-III occupancy and various genomic features or tRNA gene characteristics. We first looked at histone H3 trimethylation marks.
In RNAP-II genes, the H3K4me3 mark forms two peaks bracketing the TSS, whereas the H3K36me3 mark accumulates within the transcribed region (Barski et al. 2007; Bernstein et al. 2007; Kouzarides 2007). In human RNAP-III genes, H3K4me3 was reported to accumulate upstream of the TSS, whereas H3K36me3 was largely absent (Barski et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010). To examine the relation between these marks and RNAP-III genes in mouse liver cells, and to exclude confounding signals from nearby RNAP-II or RNAP-III transcription units, we selected tRNA genes that were removed by at least 1000 bp from the nearest RNAP-II or RNAP-III transcription unit. As shown in Figure 3A, these 206 isolated genes had a clear peak of H3K4me3 (orange line) centered from about −250 to −300 relative to the TSS, as well as a second, lower peak located downstream from the gene. The absence of H3K4me3 over the gene may reflect the absence of histone H3, as observed in human RNAP-III genes (Barski et al. 2010), and is consistent with the idea that unlike RNAP-II genes, RNAP-III genes are generally devoid of nucleosomes within the RNA coding sequence. In contrast to the H3K4me3 mark, there were no detectable H3K36me3 modifications (purple line). This was not surprising as this modification is known to be deposited onto the body of RNAP-II genes during RNAP-II elongation by a methylase binding to the RNAP-II CTD (Li et al. 2002; Krogan et al. 2003; Xiao et al. 2003; Kizer et al. 2005), a structure absent in RNAP-III.
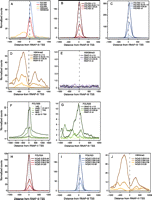
Average tag density profiles for RNAP-II and RNAP-III subunits and some histone marks. Two hundred and six tRNA genes (117 with scores <5; 26 with scores between 5 and 29.25; 28 with scores between 29.26 and 115.37; 35 with scores >115.37) removed by at least 1000 bp from the nearest RNAP-III or mRNA-encoding RNAP-II transcription unit were selected for this analysis. An analysis with genes removed by at least 1000 bp from RNAP-III and all RNAP-II transcription units including those encoding noncoding RNAs gave identical results (159 genes, 83 with scores <5; 23 with scores between 5 and 29.25; 24 with scores between 29.26 and 115.37; 29 with scores >115.37; data not shown). (A) Average tag density profile for the factors indicated on all 206 genes. (B–E,G) average tag profile for the indicated factors on genes with RNAP-III scores smaller than 5, between 5 and 29.25, between 29.25 and 115.37, and larger than 115.37, as indicated. (F) Average POLR2B tag profile on RNAP-III TSS (dotted line) and, for comparison, on RNAP-II TSS divided into quartiles with the lowest (first quartile) to the highest (fourth quartile) level of RNAP-II indicated by lines of darkening shades of green. (H–J) Tag profile for POLR3A, POLR3D, and H3K4me3, as indicated, on 206 isolated tRNA genes divided according to the percentage of CpG dinucleotides (in a region extending from 300 nt upstream of the TSS to 100 nt downstream from the RNA coding region).
We then plotted the same H3K4me3 and H3K36me3 profiles, this time separating the isolated tRNA genes into four groups as shown in Figure 3, B and C, those with scores lower than 5, those with scores between 5 and 29.25, those with scores between 29.25 and 115.37, and those with scores higher than 115.37 (see Supplemental Table S1). As shown in Figure 3, D and E, the amounts of H3K4me3 modification scaled with the amount of both POLR3A and POLR3D, whereas the H3K36me3 modification remained very low in each group. When analyzed by univariate linear regression, the correlation with H3K4me3 was strongly significant even when considering all 404 tRNA genes with scores composed of at least 80% unique tags (R2 = 0.77, P-value <2.2 × 10−16; Supplemental Table S7).
Previous data suggested that significant RNAP-II peaks were present in the vicinity of RNAP-III genes. In some case, these peaks were shown to coincide with RNAP-III genes (Barski et al. 2010; Moqtaderi et al. 2010), in others they were shown to correspond to neighboring RNAP-II TSSs (Moqtaderi et al. 2010; Oler et al. 2010). We used our set of isolated tRNA genes to avoid potentially confounding signals from neighboring RNAP-II TSSs. We detected a broad peak of POLR2B overlapping the RNAP-III TSS (Fig. 3A, green line), but as is clear in Figure 3A, where the scale is identical for all factors analyzed, the POLR2B signal was 19- and 33-fold lower than the POLR3A and POLR3D signals, respectively. Since detected amounts depend on the quality of the antibodies used for the immunoprecipitations, we compared the POLR2B signals on RNAP-III promoters with those on RNAP-II promoters. For this purpose, we used a collection of 5115 “isolated” RNAP-II genes (whose TSSs were removed from any other TSS by at least 1.5 kb) and divided them into four quartiles according to expression levels as determined by gene expression arrays (CycliX Consortium, unpubl.). Figure 3F shows the amounts of POLR2B on RNAP-II promoters in the four quartiles (lines of different shades of green) as well as the average amount of POLR2B on the isolated tRNA genes (black dotted line, corresponding to the green line in Fig. 3A). As expected, the amount of POLR2B on the RNAP-II genes scaled with the amounts of mRNAs measured by gene expression arrays. By comparison, the amount of RNAP-II detected on tRNA TSS was low, at a level intermediate between RNAP-II amounts found on the lowest and second lowest quartiles of RNAP-II genes. Nevertheless, it was clearly above background and, as shown in Figure 3G, it scaled with RNAP-III occupancy, an effect that was highly significant in univariate linear regression (R2 = 0.49, P-value <2.2 × 10−16; see Supplemental Table S7). The genes most highly occupied by RNAP-III displayed the clearest RNAP-II peak. Genes with RNAP-III scores between 29.25 and 115.37 displayed a much lower and broader RNAP-II peak, while genes with lower scores had no detectable RNAP-II. These results suggest that RNAP-III promoters are not absolutely discriminatory, and occasionally recruit RNAP-II instead of RNAP-III, in a manner proportional to RNAP-III recruitment. We did not detect significant amounts of RNAP-III on RNAP-II promoters (data not shown), suggesting that, on average, this is not the case for RNAP-II promoters.
RNAP-III-occupied isolated tRNA genes are often in regions rich in CpG dinucleotides
Highly expressed human tRNA genes are often close to RNAP-II promoters with high CpG contents (Oler et al. 2010). Here we asked whether there was any correlation between CpG content and RNAP-III occupancy at isolated tRNA genes in mouse liver. CpG content, as measured in a region extending from 300 nucleotides upstream to 100 nucleotides downstream from each tRNA gene, scaled with POLR3A and POLR3D occupancy (Fig. 3H,I), H3K4me3 (Fig. 3J), and RNAP-II occupancy (data not shown). Thus, even for isolated tRNA genes, more highly RNAP-III occupied genes tend to have a higher CpG dinucleotide content. In contrast, high G/C content was anticorrelated with RNAP-III occupancy. Both correlation with CpG content and anticorrelation with G/C content were significant in univariate linear regression (R2 = 0.36, P-value <2.2 × 10−16 and R2 = 0.08, P-value 3.0 × 10−9, respectively; see Supplemental Table S7).
RNAP-III-occupied tRNA genes are often close to active RNAP-II transcription units
The tRNA genes analyzed above are removed from other RNAP-II (and RNAP-III) transcription units. To address whether active mouse RNAP-III tRNA genes are often located close to active RNAP-II genes, as in human cells (Moqtaderi et al. 2010; Oler et al. 2010), we first examined the proximity of all 433 tRNA genes to annotated RNAP-II TSS or gene “ends” as defined by polyadenylation sites (PAS) for both coding and noncoding genes, on both strands. No enrichment in PAS was found, on either strand (data not shown). In contrast, as shown in Figure 4A, there was a significant enrichment in RNAP-II TSS near highly RNAP-III-occupied tRNA genes both on the same strand as the RNAP-III genes (Fig. 4B) as well as on the opposite strand (Fig. 4C). For both strands, 95 tRNA genes with a score larger than 29.25, but only 52 with a score smaller than 29.25, were within 5 kb of an RNAP-II TSS, (R2 = 0.13, P-value 2.0 × 10−14 in univariate linear regression for the log of the distance to the closest TSS; see Supplemental Table S7).
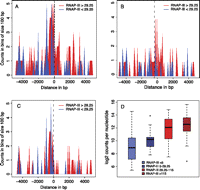
RNAP-III-occupied tRNA genes are often close to transcribed RNAP-II genes. (A) RNAP-III-occupied tRNA genes are often close to RNAP-II TSSs. The horizontal axis shows the distance separating RNAP-III and RNAP-II TSSs (0 indicates the RNAP-III TSS; negative and positive numbers indicate the regions upstream of and downstream from the TSS, respectively). The vertical axis indicates the number of tRNA genes in bins of 100 bp, separated into genes with RNAP-III occupancy scores >29.25 (red) and <29.25 (blue). (B,C) As in A, but only the RNAP-II genes transcribed in the same or opposite strand, respectively, as the tRNA gene are shown. (D) Box plots show tRNA genes with scores <5 (blue box), between 5 and 29.25 (dark blue box), between 29.26 and 115.37 (bright red box), and higher than 115.37 (dark red box). (Vertical axis) Highest number of POLR2B tags in 500-bp regions centered on an RNAP-II TSS found within 5 kb upstream of and 5 kb downstream from each tRNA gene.
We then checked whether the RNAP-II TSS close to highly transcribed RNAP-III genes were in general highly occupied by RNAP-II. We considered tRNA genes with an RNAP-II TSS within 5 Kb upstream or downstream, and plotted the amount of POLR2B tags at the most highly RNAP-II-occupied TSS within this region for tRNA genes with RNAP-III occupancy scores less than 5, between 5 and 29.25, between 29.25 and 115.37, and >115.37. As shown in Figure 4D, more highly transcribed tRNA genes tended to be close to more highly transcribed RNAP-II genes (R2 = 0.18, P-value <2.2 × 10−16 in univariate linear regression; see Supplemental Table S7). It is important to note, however, that this was only a general tendency, and that some isolated tRNA genes were highly transcribed. For example, the tRNATrp_CCA 1880 on chr11, with the 11th highest score, and the tRNAThr_ATG 1213 on chr7, with the 24th highest score, were ∼30 and 20 Kb away, respectively, from the closest annotated RNAP-II TSS.
Highly RNAP-III-occupied tRNA genes have slightly different A and B boxes from poorly or not-occupied genes
We have shown above that highly RNAP-III-occupied tRNA genes are often close to H3K4me3 modifications in CpG-rich regions, and close to RNAP-II genes actively transcribed. We then asked whether highly transcribed genes have stronger promoter elements. Figure 5A shows the LOGOS present in all tRNA genes classified by levels of POLR3A and POLR3D occupancy. The most striking feature was that A and B boxes in poorly or not transcribed genes were highly similar to those in highly transcribed genes. Nevertheless, some small differences were apparent. To characterize them better, we used the output of univariate regressions to identify bases that had significant positive or negative correlation with the scores (see Supplemental Fig. S5). We then mutated the A and B boxes of the highest score tRNA gene (chr11_1821_tRNASer_GCT-), changing each variable position to that displaying a negative correlation (Fig. 5B), and tested the effects in an in vitro transcription system. As shown in Figure 5, C and D, the mutations in each of the A and B boxes reduced transcription, although for the A box the reduction was not significant at a 5% confidence level (the P-value for the A box was 0.08990, for the B box 0.00048), and a combination of both sets of mutations led to barely detectable transcription (P-value 0.00017). Thus, even though the A and B boxes in poorly and highly transcribed genes appear very similar, the variable positions do play an important role for the overall efficiency of the promoter, especially those in the B box.
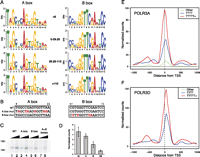
The qualities of the A and B box and terminator sequences correlate with RNAP-III occupancy. (A) The LOGOs obtained for the A and B boxes in tRNA genes with the RNAP-III scores indicated in the middle. (B) A and B box mutations introduced into the serine tRNA gene (chr11_1821_tRNASer_GCT-). Note that bases 3–6, 14, and 15 of the A box, and bases 2–6, 14, and 15 of the B box are, for most tRNAs, involved in base-pairing in the clover leaf structure. For the tRNA gene analyzed here, the bases involved in base-pairing are bases 3–5 of the A box and bases 2–6, 14, and 15 of the B box. (C) In vitro transcription with the serine tRNA gene, either wild type, or with mutations in the A, B, or both A and B boxes as indicated above the lanes. (D) Quantitation of six replicate experiments as in C. (E) Average tag density profiles for POLR3A in genes with strong terminators (5Ts or more, red line), medium terminators (4Ts, blue line), or poor terminators (less than 4Ts, green line). (F) As in E, but for POLR3D.
tRNA genes with high RNAP-III occupancy often have strong terminators
RNAP-III has the particularity of terminating transcription at runs of T residues without, at least in vitro, the help of additional factors. In human cells, many tRNA genes have a poor termination signal downstream from the tRNA coding sequence (Orioli et al. 2011). We examined whether this was the case for mouse tRNA genes, and whether this correlated with transcription efficiency. We eliminated from the analysis tRNA genes very close to each other (as in these cases, the score of a weakly occupied tRNA gene can be inflated by the presence of a large neighboring peak extending into its flanking region) and considered three classes of genes with either strong, medium, and weak or inexistent terminators (see Orioli et al. 2011). We found that out of 404 remaining mouse tRNA genes, 161 had strong terminators (five or more Ts) within 50 bp of the end of the mature RNA coding sequence, 185 had medium ones (4Ts), and 58 had weak ones or did not have any terminator within the 50-bp 3′ flanking region. Remarkably, POLR3A and POLR3D occupancy scaled with the strength of the terminator, as shown in Figure 5, E and F, and the effect was significant in univariate regression (R2 = 0.16, P-value 1.4 × 10−16). Thus, the quality of the terminator contributes to transcription efficiency and in this sense can be considered part of the promoter.
Integrated analysis of factors associated with RNAP-III occupancy at tRNA genes
The analyses presented so far revealed relations between RNAP-III occupancy and each of several individual features. One limitation of this approach is that it does not inform whether an observed relation is better explained as an indirect effect driven by a concomitant association to a third variable. To address the question, we applied multivariate regression models in which RNAP-III count density in the set of tRNA genes was predicted by several explanatory variables, such that the contribution of each variable to the prediction was adjusted by the other variables (Supplemental Table S7). The explanatory variables included were RNAP-II occupancy at RNAP-III promoters, H3K4me3 and H3K36me3 signal density, RNAP-II occupancy at nearby RNAP-II TSSs, distance to the closest RNAP-II TSS, density of CpG dinucleotides, GC content, and type of RNAP-III termination sequence (TT).
As described above, the set of some 400 tRNA genes showed very heterogeneous values of RNAP-III density, and the univariable regression models showed statistical significance (P-values ≤1%) for the correlation of each of the included explanatory variables with RNAP-III. Remarkably high values of the proportion of explained variance (R2) of RNAP-III scores were achieved by H3K4me3 (77%), RNAP-II occupancy at RNAP-III genes (49%), and by the CpG density (36%); terminator type, RNAP-II, density on nearby RNAP-II TSS, distance to RNAP-II TSSs, and GC content reached moderate values between 8% and 18%. The distance to RNAP-II TSSs and the GC content showed negative effects, that is, higher RNAP-III densities were associated with closer RNAP-II TSSs and with lower GC content.
When the variables were set in the same multivariable regression model, the effects of each variable became weaker. This was expected, as most of these variables correlate with each other, compete for predicting RNAP-III densities, and can lose statistical significance if their contribution does not improve the prediction when the other variables are already used. The explained variance of the full multivariable model was R2 = 81%, slightly higher than the highest R2 reached by one variable alone, 77% by H3K4me3, which was also the dominant factor in the multivariate model, followed by the GC content. Factors that were not significant in the multivariate regression are the H3K36me3 density and the proximity to RNAP-II TSSs. In summary, the highest RNAP-III density was generally found in those tRNA genes with an intermediate or strong termination signal located in regions with lower GC content, but high CpG density, and where the density of H3K4me3 marks and RNAP-II is higher.
Discussion
RNAP-III-occupied loci in mouse liver
We report here the analysis of RNAP-III-occupied loci in a differentiated tissue, the mouse liver. While this manuscript was under review, two papers were published that are relevant to the work described here. In the first, Kutter et al. (2011) examined the evolution of RNAP-III occupancy at tRNA genes in the liver of several mammalian species. The list of RNAP-III-occupied tRNA genes described by Kutter and colleagues is nearly identical to ours, with eight exceptions. Two tRNAGly_TCC and two tRNA Asp on chromosome 1 (lines 181, 182, 205, and 206 in Supplemental Table S1) are RNAP-III occupied in our list only, and three tRNAGly_GCC as well as one tRNAThr_TGT (lines 281, 284, 332, and 392 in Supplemental Table S1) are RNAP-III occupied in the Kutter list only. In the first four cases, the corresponding peaks are constituted exclusively of repeated tags, and would therefore not have been detected by the method used by Kutter et al (2011). As to the second set of four genes, we checked whether we could detect RNAP-III in an additional 12 ChIP-seq experiments performed with mouse liver (CycliX Consortium, unpubl.), with negative results. As in both studies, C57/BL6 males of approximately the same age (10 vs. 12–14 wk in our study) were used, these few differences remain unexplained. In the second paper, Carriere et al. (2011) looked at RNAP-III occupancy in mouse embryonic stem cells. We noticed a much lager number of differences with the tRNA genes found occupied by RNAP-III in embryonic stem cells (Carriere et al. 2011), probably due to the very different nature of the biological material, undifferentiated, pluripotent cells versus liver cells. Other differences are the absence in the Carriere study of Rmrp and Rpph1 (Supplemental Table S3, lines 8,9) as well as all of the Rn5s genes (Supplemental Table S2), in this last case probably due to the repeated nature of the tags, and the presence of three Rn5s-like sequences, two Bc1 loci, and seven Rn4.5s loci between positions 47,600,661 and 47,728,438 on chromosome 6, for which we obtained no or very unconvincing (three of the Rn4.5s sequences) peaks (Supplemental Table S4). Moreover, there are many differences in occupied SINEs and uncharacterized loci, resulting either from biological or experimental differences between the two studies.
When comparing our mouse data with the RNAP-III loci we previously found occupied in human cultured cells, we find many similarities. Noticeably, although RNAP-III genes are located in genomic regions that are nearly always labeled as complex rearrangements in the chained human–mouse alignments defined in the UCSC genome tables (Chiaromonte et al. 2002; Kent et al. 2003; Schwartz et al. 2003), we could assign shared synteny relationships to some 229 tRNA genes, considering only pairs of identical isoacceptors at similar locations relative to RNAP-II and other RNAP-III transcription units. Moreover, for all other known RNAP-III transcription units (15 genes) listed in Supplemental Table S3, human–mouse shared synteny relationships could be clearly established. When looking at this 244 RNAP-III gene subset, we find that the scores are linked. This is remarkable if one considers that we are comparing human culture cells (IMR90Tert cells) and mouse liver cells in experiments with tags sequenced at 35 nt in the first case and 76 nt in the second case. On the other hand, the moderate correlation is consistent with the recent systematic comparison of RNAP-III binding at tRNA genes in six mammals, which reveals both conservation and differences of RNAP-III binding at tRNA genes with shared synteny (Kutter et al. 2011). Interestingly, Kutter et al. (2011) find, in addition, that when comparing total RNAP-III occupancy of all isoacceptor tRNA genes belonging to a certain isotype family, there is very strong conservation among different mammalian species.
We did not explore in detail possible shared synteny relationships between RNAP-III-occupied mouse and human SINEs, but one particular example stood out. The most highly RNAP-III-occupied SINE in the mouse liver was a MIR in the first intron of the Polr3e gene, which encodes an RNAP-III-specific subunit. The MIR is transcribed in the opposite direction of the RNAP-II Polr3e gene. We observed previously the very same arrangement in human cells, where a MIR within the first intron of the human POLR3E gene, and transcribed in the opposite direction, was occupied by RNAP-III and some of its transcription factors (see Supplemental Table S5, line 48 in Canella et al. 2010). This striking similarity indicates conservation of both MIR location and MIR RNAP-III promoter activity, and thus suggests function. One could imagine that the antisense RNA transcribed by RNAP-III regulates in some way the activity of the Polr3e gene or the stability of the pre-mRNA. However, we favor the possibility that it is, in fact, the process of antisense RNAP-III transcription itself, rather than the resulting RNA product, that regulates the Polr3e output. Indeed, the RNAP-II peak observed at the 3′ end of the MIR reflects RNAP-II accumulation at this specific location, a phenomenon not commonly observed within the body of RNAP-II transcription units. It is suggestive of an elongation block at this location. Thus, the patterns of RNAP-II and RNAP-III accumulation on this transcription unit are consistent with active antisense RNAP-III transcription creating a roadblock to RNAP-II transcription. This may thus constitute an example of RNAP-III transcription influencing RNAP-II transcription efficiency. There may be other examples, as several of the SINEs transcribed in mouse (and human) cells are located within introns of RNAP-II genes. Although they are generally transcribed on the same strand as the RNAP-II gene, they may also constitute roadblocks to RNAP-II transcription. Incidentally, the RNAP-II peak at the 3′ end of the MIR indicate that the very same template can be transcribed on both strands concomitantly, a fact that has been difficult to establish.
Links between RNAP-III and RNAP-II transcription
Several previous lines of evidence have suggested a link between RNAP-II and RNAP-III transcription. One is the observation that in the case of the human RNU6 snRNA genes, RNAP-II peaks between 300 and 600 nt upstream of the RNU6 TSS, depending on the RNU6 gene (Listerman et al. 2007). Based in part on effects of α-amanitin treatments, it has been suggested that these peaks represent actively transcribing RNAP-II, and that this active transcription is required for efficient RNAP-III transcription at the RNU6 genes (Listerman et al. 2007). We checked whether we could detect accumulation of RNAP-II upstream of the five Rnu6 snRNA genes, as well as the Rnu6atac gene in mouse liver cells. There was, indeed, often a small accumulation of RNAP-II upstream of the mouse Rnu6 snRNA genes, but there was no correlation between these RNAP-II peaks and the amount of RNAP-III on the downstream Rnu6 gene. For example, the Rnu6 gene with the lowest RNAP-III score, on chromosome 9 (see Supplemental Table S3), had about the same amount of RNAP-II in its 5′-flanking region as the Rnu6atac gene, which had the highest score of the Rnu6 genes, and the Rnu6 gene with the second highest RNAP-III score had no significant peak of RNAP-II in its 5′-flanking region (data not shown). Thus, the patterns observed in mouse cells do not support the idea that RNAP-II transcription in the −600 to −300 region of Rnu6 genes contributes to high RNAP-III transcription of the Rnu6 genes.
The other observations linking RNAP-II and RNAP-III indicate that in human cells, active RNAP-III genes are often close to RNAP-II TSSs, and to peaks of RNAP-II occupancy. Such peaks of RNAP-II occupancy have been reported to occur either at (Barski et al. 2010) or in the vicinity of (Moqtaderi et al. 2010; Oler et al. 2010) RNAP-III genes. Cross-linking of RNAP-II at RNAP-III genes suggests that RNAP-III promoters may sometimes recruit RNAP-II instead of RNAP-III, whereas cross-linking of RNAP-II in the vicinity of RNAP-III genes may represent RNAP-II recruitment to nearby RNAP-II TSSs. To distinguish between these possibilities, we examined a set of “isolated tRNA genes” removed from any other annotated RNAP-II or RNAP-III transcription unit by at least 1000 bp. With this set of genes, we could clearly show that there is in fact some RNAP-II present at tRNA genes in mouse cells, and the amount of RNAP-II scales with the amount of RNAP-III. For tRNA genes moderately occupied by RNAP-III (second tertile), RNAP-II appears to be recruited over a broad region upstream of the TSS, through a mechanism that remains unexplained. For the tRNA genes most highly occupied by RNAP-III, the RNAP-II peak is at or very close to the TSS, strongly suggesting that very active RNAP-III promoters do, with a certain frequency, recruit the “wrong” polymerase, i.e., RNAP-II. The levels are low, however, with the most active tRNA genes recruiting amounts of RNAP-II comparable to those recruited by RNAP-II genes in the second lowest activity quartile (see Fig. 3F,G). tRNA promoters are thus not absolutely selective in which type of polymerase they recruit. Carriere et al. (2011), examining all RNAP-III-occupied tRNA genes, observed RNAP-II (largely hypophosphorylated) in a broad region upstream of the genes, perhaps corresponding to the broad peak that we observe for tRNA genes of the second tertile, but no clear occupancy on the gene itself. The difference may be due to our use of isolated tRNA genes. In any case, it will be interesting to determine whether highly active RNAP-III tRNA genes can be transcribed by RNAP-II in vitro.
Separate from the presence of RNAP-II at RNAP-III genes, we found in univariate regressions that RNAP-III-occupied tRNA genes are often close to RNAP-II TSSs, and that they are often close to RNAP-II TSSs displaying peaks of RNAP-II. In the multivariate regression analysis, both of these correlations became non- or close to nonsignificant (Supplemental Table S7). This may reflect that the presence of RNAP-II at RNAP-II TSSs close to RNAP-III genes is, in fact, not directly correlated to RNAP-III occupancy at tRNA genes, but rather to a third variable such as the presence of the H3K4me3 mark, which is itself correlated to RNAP-III occupancy. Indeed, as reported for human cells (Barski et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010), we find that in mouse liver cells, the H3K4me3 mark occurs upstream of as well as downstream from RNAP-III genes, with a strong depletion over the gene itself, even for isolated tRNA genes, suggesting that the H3K4me3 mark is delivered independent of nearby RNAP-II promoters. Indeed, as illustrated in Supplemental Figure S6, there is a fairly high proportion of tRNA genes with abundant RNAP-III signal, yet low RNAP-II, but much fewer with abundant RNAP-III signal, yet low H3K4me3 signal.
Promoter and terminator sequences
Examination of the A and B box sequences revealed very few differences between highly and poorly expressed tRNA genes. Nevertheless, changing all highly variable positions to the nucleotides found most often in poorly expressed tRNA genes significantly reduced transcription in vitro, especially for the B box and the combination of A and B box mutations. Thus, the variable positions in the A and B boxes are likely to play a direct role in transcription efficiency in vivo. We also noticed that highly RNAP-III-occupied tRNA genes generally have a very strong terminator consisting of five or more Ts, whereas less RNAP-III-occupied genes have weaker terminators. It has previously been shown that transcription efficiency of the Ad2 VA1 gene in vitro diminishes as the VA1 terminator is replaced with weaker terminators (Wang et al. 2000). Further, RNAP-III has been shown to recycle preferentially on the same DNA template in vitro, and this “facilitated recycling” is dependent on the terminator (Dieci and Sentenac 1996). Thus, higher RNAP-III occupancy at genes with strong terminators may result from more efficient recycling of the enzyme.
We were able to incorporate the terminator variable into the multivariate linear model, but not the information about the sequence of the A- and B-box. Indeed, because almost all single nucleotide variants were fairly rare, the effects of single-point mutations could not be well estimated, and, in fact, the addition of these variables did not improve the prediction of RNAP-III density. It seems likely, however, that a large part of the 19% variability not explained by the linear model derives from the exact sequence of the A and B boxes.
The analysis of the mouse RNAP-III-occupied loci reveals a genomic organization of RNAP-III genes that is quite similar to that found in the human genome, as shown by shared synteny relationships. It shows that active RNAP-III genes are in open chromatin regions, and that weakly and strongly transcribed RNAP-III genes have different terminator and promoter sequences. The model that emerges from this analysis suggests that the underlying genomic organization dictates chromatin organization and influences RNAP-III transcriptional activity. RNAP-III transcription efficiency can be influenced both by this chromatin organization and by the quality of the promoter and terminators of individual genes.
Methods
Animals
C57/BL6 male, 12–14-wk-old (at time of sacrifice), mice were entrained with a 12h light/12h dark light regimen with food access between ZT12 and ZT24 for 7 d (ZT0 is defined as the time when the lights are turned on and ZT12 as the time when the lights are turned off). At each ZT02 and, as a biological replicate, ZT26, five mice were anesthetized with isoflurane and decapitated. The livers were perfused with 5 mL of PBS through the spleen and immediately collected. Up to 100 mg of liver was snap-frozen in liquid nitrogen and kept at −80°C for RNA extraction. The rest of the livers were immediately homogenized in PBS containing 1% formaldehyde for chromatin preparation. All animal care and handling was performed according to the State of Geneva's law for animal protection.
Chromatin immunoprecipitations
Perfused livers were processed for chromatin preparation as described in Ripperger (2006). The chromatin samples from the five mice were pooled, frozen in liquid nitrogen, and stored at −70°C. For the ChIP, the following antibodies were used: anti-POLR2B (Santa Cruz Biotechnology, H-201), anti-POLR3D (CS681, raised against the same peptide as CS682) (see Chong et al. 2001), anti-POLR3A (CS377) see (Sepehri and Hernandez 1997), anti-H3K4me3 (Abcam, ab8580), and anti-H3K36me3 (Abcam, ab9050). To determine the optimal amounts of each antibody, we performed test ChIPs and determined enrichment for a set of promoters by real time PCR. Further details are provided in the Supplemental Methods.
RNA isolation and analysis
About 100 mg of snap-frozen liver tissue was disrupted in 1 mL of TRIzol reagent (Invitrogen) with a GentleMACS homogenizer (Miltenyi Biotec) and centrifuged for 10 min at 12,000g at 4°C. The cleared homogenate solution was incubated at room temperature for 5 min. A total of 200 μL of chloroform was then added, the samples were inverted several times and centrifuged for 15 min at 12,000g at 4°C; 1.5 vol of 100% ethanol was then added to 100 μL of the recovered aqueous phase and the RNA was purified with a miRNeasy Mini Kit (Qiagen) following the manufacturer's instructions. The rest of the aequous phase (∼200 μL) was kept frozen. For each ZT, 500 ng of total RNA from each of the five mouse livers were pooled and hybridized to Mouse Gene 1.0ST arrays (Affymetrix).
Total RNA quantities were assessed with a NanoDrop ND-1000 spectrophotometer and the RNA quality was assessed on RNA 6000 NanoChips with the Agilent 2100 Bioanalyzer (Agilent Technologies). For each sample, 100 ng of total RNA was amplified with the WT Expression kit (Invitrogen; catalog no. 4411974); the resulting sense cDNA was fragmented with uracil DNA glycosylase (UDG) and apurinic/apyrimidic endonuclease 1 (APE-1), and biotin-labeled with terminal deoxynucleotidyl transferase (TdT) using the GeneChip WT Terminal labeling kit (Affymetrix; catalog no. 900671). Affymetrix Mouse Gene 1.0 ST arrays were hybridized with 2.5 μg of biotinylated target at 45°C for 17 h, washed, and stained according to the protocol described in Affymetrix GeneChip Expression Analysis Manual (Fluidics protocol FS450_0007).
The arrays were scanned with an Affymetrix GeneChip Scanner 3000 7G, and the raw data was extracted from the scanned images and analyzed with the Affymetrix Power Tools software package.
All statistical analyses were performed with the statistical language R and various Bioconductor packages (http://www.Bioconductor.org). Hybridization quality was assessed with the Expression Console software (Affymetrix). Normalized expression signals were calculated from Affymetrix CEL files using the RMA normalization method. Differential hybridized features were identified using the Bioconductor package “limma,” which implements linear models for microarray data (Smyth 2004). P-values were adjusted for multiple testing with Benjamini and Hochberg's method to control the false discovery rate (FDR) (Benjamini and Hochberg 1995).
Analysis of ChIP-seq data
Tag alignment
Seventy-six nucleotide long sequence tags were aligned essentially as described previously (Canella et al. 2010). Further details are provided in the Supplemental Methods.
Peak detection
In a first “gene discovery” step, we used the sissrs software (http://www.rajajothi.com/sissrs/) (Jothi et al. 2008), modified to take into account both tags with unique and multiple matches in the genome for peak detection as described previously (Canella et al. 2010). Further details are provided in the Supplemental Methods.
Score calculation
The scores for the retained loci as well as all annotated tRNA genes, whether or not showing sissrs peaks (loci in Supplemental Tables S1–S5), were then calculated by adding all tags covering the RNA coding region as well as 150 bp upstream of and downstream from the RNA-coding sequence and dividing the resulting number by the length of the region. In cases where the distance separating two genes was shorter than 300 bp, we divided the distance into two equal parts and attributed the tags in each part to the closest gene. Each score could have two components: one was the sum of tags with unique matches in the genome, the other represented tags with multiple matches in the genome. Such tags were attributed a weight corresponding to the number of times they were sequenced divided by the number of matches in the genome, with a maximum weight set at 1. The cut-offs used were established as follows: We computed signal scores over 50,000 regions chosen at random on the genome. The random regions were 371 bp long, the typical length of a tRNA gene plus the 150-bp extensions on the 5′ and 3′ ends. The distribution of scores is shown in Supplemental Figure S7. We found 37 regions that had a score higher or equal to 5 (P-value 0.00074), and 50 regions with scores higher or equal to 4.54 (P-value 0.001). We chose the cut off at a P-value of 0.001 and rounded this number to 5. The rest of the RNAP-III-occupied features was divided into three tertiles, leading to classes with scores of 5–29.25, 29.26–115.37, and larger than 115.37.
Use of input data
For both the Rep1 and Rep2 sample, we sequenced the chromatin input after decross-linking. The total number of aligned tags obtained is shown in Supplemental Table S8 and is in excess of 25 million. We then computed the number of tags sequenced in the input material and aligning over each scored region. As shown in Supplemental Figure S8A, this number is very low with most genes, which can be as short as 70–80 bp for a typical tRNA gene, displaying fewer than eight tags in the input material. (By comparison, the number of tags obtained in the anti-RPC1 or anti-RPC4 immunoprecipitations over the same regions could reach more than 2000; Supplemental Fig. S8B). This suggests that the number of input tags in these regions does not reflect some biological property (such as open chromatin), but rather random noise. Indeed, dividing the scores by the amount of tags in the input (in log2 space) led to a decreased correlation between replicates. The decrease in correlation observed when dividing scores by input could reflect a common bias present in both replicates, although the low correlation between H3K36me3 and any other ChIP-seq count variable argues against it, as a strong bias should introduce correlation between all of the variables. Nevertheless, we repeated the univariate and multivariate regressions, as well as some of the figures quantifying the data with log ratios as follows: log [(y+1)/(i+c)], where y = ChIP score, i = input score, and c = pseudocounts and is varied c = 5, 10. As can be seen by comparing Supplemental Table S7 above with Supplemental Table S9 (c = 5) and Supplemental Table S10 (c = 10) showing the numbers obtained with the second method, the strong effects (highlighted in yellow in the tables) (strong correlation with H3K4me3 and CpG content) remained close to identical, and the R2 value and P-value for the full multivariate model were identical. All other effects showed only small variations. We also repeated the analyses in Figure 3 (D and G), as shown in Supplemental Figure S9, with similar results. Thus, as expected, all conclusions are very insensitive to the choice of approach for analysis.
In vitro transcription
In vitro transcription (IVT) experiments were performed as in Lobo et al. (1992). Briefly, IVT reactions were conducted in 10 mM HEPES (pH 7.9), 5% glycerol, 100 mM KCl, 0.1 mM EDTA, 1 mM spermidine (Sigma), 1 mM DTT, 5 mM MgCl2, 1 mM each ATP, UTP, CTP, and 10 μC of [α-32P]GTP (500 Ci/mmol) in a total volume of 20 μL containing 20–30 μg of whole-cell extract.
Data access
The data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE33421.
The CycliX Consortium
Primary investigator
Nouria Hernandez9
Co-primary investigators
Mauro Delorenzi,10 Bart Deplancke,11 Béatrice Desvergne,9 Nicolas Guex,12 Winship Herr,9 Felix Naef,11 Jacques Rougemont,13 and Ueli Schibler14
Bioinformatics coordination
Nicolas Guex12
Animal breeding, conditioning, collections of biological material
Teemu Andersin,14 Pascal Cousin,9 Federica Gilardi,9 Pascal Gos,14 Gwendal Le Martelot,14 and Fabienne Lammers9
Chromatin immunoprecipitations, libraries, gene expression arrays
Computing infrastructure installation and servers maintenance
Roberto Fabbretti,12 Arnaud Fortier,12 Li Long,12 Volker Vlegel,12 and Ioannis Xenarios9,10,12
Tag mapping, quantification, and normalizations
Eugenia Migliavacca,12 Viviane Praz,9 Nicolas Guex,12 Felix Naef,11 and Jacques Rougemont13
Data management and viewing tools
Fabrice David,10,13 Yohan Jarosz,10,13 Dmitry Kuznetsov,12 Robin Liechti,12 Olivier Martin,12 Frederick Ross,10,13 and Lucas Sinclair10,13
Acknowledgments
We thank Marianne Renaud for invaluable help with the preparation of the manuscript. We thank Keith Harshman, Director of the Lausanne Genome Technologies Facility, where all of the ultra-high-throughput sequencing was performed, and Ioannis Xenarios, Director of the Vital-IT (http://www.vital-it.ch) Center for High Performance Computing of the Swiss Institute of Bioinformatics. Maintenance of the CycliX servers was provided by Vital-IT. This work was financed by CycliX, a grant from the Swiss SystemsX.ch initiative evaluated by the Swiss National Science Foundation, Sybit, the SystemsX.ch IT unit, the University of Lausanne, the University of Geneva, the Ecole Polytechnique Fédérale de Lausanne (EPFL), and Vital-IT.
Footnotes
-
↵7 A complete list of consortium authors appears at the end of this manuscript.
-
↵8 Corresponding authors.
E-mail mauro.delorenzi{at}unil.ch.
E-mail nouria.hernandez{at}unil.ch.
-
↵9 Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, 1015 Lausanne, Switzerland.
-
↵10 Swiss Institute of Bioinformatics, University of Lausanne, 1015 Lausanne, Switzerland.
-
↵11 Interfaculty Institute of Bioengineering, School of Life Sciences, Ecole polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland.
-
↵12 Vital IT, Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland.
-
↵13 Bioinformatics and Biostatistics Core Facility, School of Life Sciences, Ecole polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland.
-
↵14 Department of Molecular Biology, Faculty of Sciences, University of Geneva, 1211 Geneva, Switzerland.
-
↵15 Département de formation et de recherche, Centre Hospitalier Universitaire Vaudois and University of Lausanne, 1011 Lausanne, Switzerland.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.130286.111.
- Received August 5, 2011.
- Accepted December 6, 2011.
- Copyright © 2012 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








