
Promoter architecture of mouse olfactory receptor genes
- Charles Plessy1,14,
- Giovanni Pascarella2,3,12,14,
- Nicolas Bertin1,14,
- Altuna Akalin4,10,14,
- Claudia Carrieri2,
- Anne Vassalli5,11,
- Dejan Lazarevic2,3,6,
- Jessica Severin1,
- Christina Vlachouli2,
- Roberto Simone2,3,13,
- Geoffrey J. Faulkner7,
- Jun Kawai1,
- Carsten O. Daub1,
- Silvia Zucchelli2,3,8,
- Yoshihide Hayashizaki1,
- Peter Mombaerts5,9,
- Boris Lenhard4,15,
- Stefano Gustincich2,3,15 and
- Piero Carninci1,15
- 1RIKEN Yokohama Institute, Omics Science Center, Yokohama, Kanagawa 230-0045, Japan;
- 2International School for Advanced Studies, Sector of Neurobiology, Trieste 34136, Italy;
- 3The Giovanni Armenise–Harvard Foundation Laboratory, Sector of Neurobiology, International School for Advanced Studies, Trieste 34136, Italy;
- 4University of Bergen, Bergen Center for Computational Science–Computational Biology Unit and Sars Centre for Marine Molecular Biology Bergen, 5008, Norway;
- 5The Rockefeller University, New York, New York 10065, USA;
- 6Cluster in Biomedicine, Trieste 34149, Italy;
- 7Roslin Institute and Royal (Dick) School of Veterinary Studies, The University of Edinburgh, Roslin EH25 9RG, United Kingdom;
- 8Italian Institute of Technology–International School for Advanced Studies Unit, Trieste 34136, Italy;
- 9Max Planck Institute of Biophysics, Frankfurt 60438, Germany
-
↵14 These authors contributed equally to this work.
Abstract
Odorous chemicals are detected by the mouse main olfactory epithelium (MOE) by about 1100 types of olfactory receptors (OR) expressed by olfactory sensory neurons (OSNs). Each mature OSN is thought to express only one allele of a single OR gene. Major impediments to understand the transcriptional control of OR gene expression are the lack of a proper characterization of OR transcription start sites (TSSs) and promoters, and of regulatory transcripts at OR loci. We have applied the nanoCAGE technology to profile the transcriptome and the active promoters in the MOE. nanoCAGE analysis revealed the map and architecture of promoters for 87.5% of the mouse OR genes, as well as the expression of many novel noncoding RNAs including antisense transcripts. We identified candidate transcription factors for OR gene expression and among them confirmed by chromatin immunoprecipitation the binding of TBP, EBF1 (OLF1), and MEF2A to OR promoters. Finally, we showed that a short genomic fragment flanking the major TSS of the OR gene Olfr160 (M72) can drive OSN-specific expression in transgenic mice.
In rodents, olfaction initiates mainly in the main olfactory epithelium (MOE). The recognition of volatile odorants occurs on the surface of the cilia of olfactory sensory neurons (OSNs), where olfactory receptors (ORs) are located. The extraordinary chemical diversity of olfactory ligands is matched in the mouse genome by a repertoire of more than 1100 intact OR genes encoding for G-protein-coupled receptors (GPCRs) (Buck and Axel 1991; Zhang et al. 2007). ORs can be phylogenetically categorized into two classes: Class I receptors, which have counterparts in the whole vertebrate lineage, and Class II receptors, which are specific for tetrapods (Glusman et al. 2001; Nei et al. 2008).
Each mature OSN in the MOE is thought to express only one allele of a single OR gene—monoallelic and monogenic expression, respectively. The population of OSNs that express a given OR is relatively small and can differ over two orders of magnitude (Nei et al. 2008). A given OR gene is expressed in a mosaic or punctate pattern of OSNs within a characteristic zone of the MOE. Axons of OSNs that express the same OR gene coalesce into one or a few glomeruli of the olfactory bulb (OB).
The transcriptional mechanisms that underlie this extraordinary restriction in gene expression remain unclear.
While some mammalian OR promoters have been reported to feature a conserved TATA-box (Bulger et al. 2000), others have been described as TATA-less (Sosinsky et al. 2000). They feature two well-established cis-regulatory elements: EBF1 (also known as OLF1) sites (EBF-like sites), which have been identified in the promoters of a few OR genes and also in several other OSN-specific genes, and homeodomain binding sites (HD sites), which are located in the proximity of the EBF-like sites. These two TF binding site motifs have been implicated experimentally in OR genes' expression by site-directed mutagenesis in vivo (Rothman et al. 2005). The HD site in the promoter region of Olfr151 (M71) can be bound by LHX2, a LIM-homeobox protein that is required for the expression of Class II OR genes and/or the maturation of OSNs that express this class of receptors (Hirota and Mombaerts 2004; Kolterud et al. 2004; Hirota et al. 2007).
Historically, the major limitation to the understanding of transcriptional regulation for OR genes has been the lack of a proper characterization of OR promoters and transcription start sites (TSSs). For most OR genes, only the predicted coding sequence had been annotated in the genome (Zhang and Firestein 2002), calling for the identification of the TSSs throughout the OR gene repertoire. Indeed, a custom tiling microarray data set has recently been used to study the structure of promoters for 86% of mouse OR intact genes (Clowney et al. 2011). This approach is based on a custom platform exclusively focused on the olfactory genome and relies on probes generated by RLM-RACE using degenerated primers specific for OR transcripts. Although effective for locating OR 5′ UTRs, such an idea-tailored platform lacks single-nucleotide resolutions, and its design excludes information about antisense transcription, noncoding RNAs, and non-OR genes networks in the MOE, each of these elements being potentially critical for the formulation of comprehensive models for monoallelic and monogenic expression of OR genes.
We have previously developed the cap analysis gene expression (CAGE) technology for the systematic study of transcription starting sites (TSSs) in eukaryotic cells and tissues (Carninci 2009). CAGE is based on sequencing the 5′ ends of mRNAs, of which the integrity is inferred by the presence of their cap. The sequences—which we refer here to as “tags”—are sufficiently long to be aligned in most cases at a single position of the genome. The first position of such an alignment identifies a base pair where transcription is initiated; we refer to it as a TSS in the sense of the sequence ontology's term SO:0000315. Furthermore, counting the number of times a given tag is represented in a library gives an estimate of the expression level of the corresponding transcript. CAGE has enabled us to map the transcription factor binding sites (TFBSs) in promoters (SO:0000167) (Carninci et al. 2006) and to identify long interspersed nuclear elements (LINEs) as a source of alternative promoters for protein-coding genes (Faulkner et al. 2009). To expand this approach to tiny amounts of ex vivo tissue, we have recently developed nanoCAGE, a technology that miniaturizes the requirement of CAGE for starting RNA quantity to the nanogram range and that can also be applied to RNA obtained from fixed tissues (Plessy et al. 2010).
Here, we have applied nanoCAGE technology to characterize extensively the transcriptome and the active promoters of the mouse MOE, and to elucidate the 5′ structure of the OR genes. We isolated the MOE by laser capture microdissection (LCM) from fixed histological sections of C57BL/6J mice. Using deep sequencing, we collected ∼53 million sequence tags and aligned 78% of these to unique sites in the mouse genome. We grouped these tags to identify TSSs and promoters and to quantify the expression of 955 (87.5% of 1092) OR genes and sense and antisense transcripts in the MOE. Bioinformatic analysis of genomic regions centered on promoters of OR genes yielded a map of TFBSs enrichment, and in vivo experimental validation revealed transcription factors and new candidate regulatory elements that may be involved in OR gene regulation.
Results
The transcriptional landscape of the mouse MOE
Using zinc-fix as an optimal fixative for both tissue morphology and RNA integrity preservation, adjacent histological cryosections were prepared from the MOE of C57BL/6J mice at 12 or 20–22 d postnatally. The MOE was isolated by LCM from these sections (Supplemental Fig. S1). After RNA purification, two independent nanoCAGE libraries were synthesized. The reaction was random-primed in order to target also noncoding, nonpolyadenylated RNAs [poly(A)−], and long, partially degraded RNAs. After cDNA amplification, 25-base tags were sequenced on an Illumina GA sequencer. A total of 53,158,862 tags were obtained, of which 41,399,873 (78%) could be aligned to the mouse genome.
The aligned tags were then associated to the transcript models from the reference sequence collection (RefSeq; http://www.ncbi.nlm.nih.gov/RefSeq/) and from the FANTOM3 full-length noncoding RNAs data set (Carninci et al. 2005). Conservatively, we counted a tag as evidence for the expression of a given transcript if it aligned to its 5′ UTR or to its proximal promoter, which we define as the region up to −500 bases upstream of the 5′ end of the transcript. The expression value of tags that can align to multiple loci was distributed between them according to the weighting strategy of Faulkner et al. (2008) (see Methods) and then normalized by dividing the tag counts by the total number of tags, resulting in values expressed in tags per million (tpm).
Our previous analysis with CAGE has shown that promoters can vary in shape, with some genes having a strong preference for one particular base pair for transcription initiation, and others using a broad collection of TSSs within a region of approximately 100 bases. In both cases, we clustered TSSs together when they map within 20 bases from each other, because this distance has been found effective to group together TSSs that belong to the same promoter. The TSS with the highest tag count in these clusters is chosen as the major TSS (SO:0001238) of the promoter (Carninci et al. 2006).
Twenty-six percent of the tags aligned within 200 bases of the 5′ end of a transcript (Fig. 1A,B). Fifty-two percent of the tags are associated with coding transcripts, and 19% with noncoding transcripts. Interestingly, as many as 13% of the tags are associated with the opposite strand of known transcripts (Fig. 1C), confirming that divergent transcription is a common phenomenon in mammalian promoters (Engström et al. 2006). Ten percent of the tags are mapped over the proximal promoter of coding and noncoding RNAs (6.7% and 3.3%, respectively). Furthermore, 10.7%, 11.2%, and 12.2% are found, respectively, in the 5′ UTRs, coding sequence, and 3′ UTRs of coding gene models (Fig. 1C). Twelve percent of the tags aligning to the genome are not associated with any transcript and may represent the TSSs of genes that have yet to be characterized. In addition, we noted that 10% of the tags mapped to repeat elements, a fraction that corresponds to the proportion observed in mouse brain tissues (Faulkner et al. 2009), with a predominance of SINEs (1.8%), LINEs (1.7%), simple repeats (1.2%), and LTR families of repeats (1%).

MOE transcription start sites recapitulate known transcript initiation and reveal the extent of noncoding transcripts. (A) The distribution of distances between nanoCAGE TSSs and the closest documented 5′ end of a transcript (RefSeq model or full-length noncoding FANTOM3 RNA). The number of CAGE tags is represented on different logarithmic scales for sense and antisense directions. The areas within 200 bp or 1000 bp are shaded in, respectively, darker or lighter shades of red (for TSS located on the same strand), or green (for TSS located on the opposite strand). (B) Proportion of TSSs located <200 bp (dark color), <1000 bp (light color), or >1000 bp (white) from RefSeq or FANTOM3 noncoding RNA transcripts, on the sense (red) or antisense (green) strand, both of which correspond to the distribution plotted in panel A. The white area completing the histograms depicts the remaining proportion of nanoCAGE TSS distant of >1000 bp from those documented transcripts' 5′ ends. (C) Histogram depicting the proportion of tags aligned to the proximal promoter of transcript models (defined as the region spanning from the 5′ end to 500 bp upstream), the 5′ UTR, the coding sequence (CDS), the 3′ UTR (in decreasing purple colors), the proximal promoter of FANTOM3 noncoding RNA (in orange), and the FANTOM3 noncoding RNA (in light orange). The upper part of the bar plot shows TSSs located on the same strand as the annotation, while the lower part depicts TSSs located on the opposite strand. (Gray bar) The percentage of TSSs that do not colocalize with any of those annotations.
Comprehensive map of mouse OR promoters
The absence of a genome-wide definition of TSSs for OR genes is a great obstacle to the identification of transcription factors (TFs) that may be involved in the regulation of their expression. Therefore, we analyzed the nanoCAGE tags that map to the putative promoter-containing regions of OR genes. Of these, 606,260 tags (1.5% of the MOE transcriptome) are located in evolutionarily conserved clusters of OR genes (CLICs) (Supplemental Table S1; Aloni et al. 2006). These tags are aggregated in 26,452 individual TSSs.
In contrast with the broad promoter shape of widely expressed transcripts, 88.5% of OR promoters are of a sharp type with only a single dominant TSS position, a known feature of tissue-restricted transcripts (Carninci et al. 2006). Moreover, 21% of OR promoters have a canonical TATA-box (Ponjavic et al. 2006). Inspection of the limited number of previously cloned OR 5′ UTRs (Lane et al. 2002; Michaloski et al. 2006) reveals that they are conserved in orthologous loci in the rat and often in the dog genome. Using sequence conservation and expression level for selection, and after removing tags corresponding to expressed repeat elements in order to avoid accidental assignment of a non-OR promoter to an OR gene, we assigned one cluster of nanoCAGE TSSs as the promoter for 955 (87.5% of 1092) mouse OR genes with an intact open reading frame (Supplemental Table S2).
The normalized expression levels of the 955 OR promoters were distributed between 0.02 and 142.2 tpm (first quartile: 0.70; median 2.17; third quartile: 5.05). The alignment of 87% of the tags to OR promoters was unique. Accordingly, the OR expression profiles obtained with or without including the tags aligning in multiple loci were almost identical (Pearson coefficient: 0.998). The expression level of each OR promoter is available in the Supplemental Material.
Most of OR promoters are several thousand bases away from the closest annotated 5′ boundary of OR transcript models, which in most cases is merely the start of the coding sequence, in agreement with observations that OR genes typically have a noncoding first exon (Lane et al. 2002; Michaloski et al. 2006). The median distance between the transcription and translation start sites is 3125 bases (first quartile: 1926; third quartile: 4890).
The most comprehensive OR TSS analysis before we started our study consisted of a 5′-PCR-based analysis of 198 mouse OR genes (Michaloski et al. 2006). We assigned these 5′-RACE products to OR genes. Eighty-eight percent had a promoter determined by nanoCAGE. Since 5′ RACE also has a single-nucleotide resolution, we compared the relative position between each RACE EST and their corresponding nanoCAGE major TSS. The median distance was 26 bases. In addition, we also validated the 5′ ends of transcripts for Olfr2 (MOR103-15), Olfr329 (MOR275-6P), and Olfr1215 (MOR233-13) with RACE and capillary sequencing. Lastly, we compared our TSS with the ones defined by hybridization of RLM-RACE-PCR products on tilling arrays by Clowney et al. (2011) and found that their median directed distance was of −69 nt (Supplemental Fig. S2). This systematic shift might be introduced at the step where the hybridization signal of the tiling arrays is transformed into transcript boundaries, because this involves determining detection thresholds.
Together our OR promoters comprised 20% of the tags mapping onto evolutionarily conserved clusters of OR genes (CLICs) (Aloni et al. 2006). Twenty-one percent of the tags in CLICs were associated to non-OR RefSeq transcripts. While 45% of the tags mapping to CLICs aligned in intergenic regions, defined by RefSeq complemented with our OR promoters, we note that six out of the 10 most expressed intergenic loci in CLICs were supported by FANTOM3 CAGE tags or mouse mRNAs deposited in GenBank, suggesting that CLICs contain more non-OR genes than previously reported. Seven percent of the tags mapped to OR CDS, and 4% between the OR promoters and translation start site, revealing potential alternative transcriptional start sites for 662 of the 955 OR genes for which we assigned a promoter. Expressed LINE, LTR, and satellite repeats (but not SINE or simple repeats families) were significantly over-represented in CLICs (Fisher test P-value < 10−10; see Methods for details). More than 88% of them were located in intergenic regions.
3′ UTR and antisense transcripts at OR gene loci
In addition to the TSSs for protein-coding OR transcripts, we identified at OR gene loci a large number of TSSs previously uncharacterized. Former reports showed the prevalence of 3′ transcripts comprising only part of the 3′ UTR of coding loci (Carninci et al. 2006; Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project 2009). In line with these results, we found TSSs directly 3′ to the coding sequence of OR genes. Examples are shown for an OR gene cluster on chromosome 7 (Fig. 2). Furthermore, we discovered antisense transcripts in 505 OR loci; 276 of these overlap with the coding sequence. The distribution of antisense transcripts is similar across Class I and II OR genes. We validated by random-primed 3′ Deep-RACE the presence of antisense transcripts at Olfr49 (MOR118-1), Olfr522 (MOR103-5), and Olfr702 (MOR260-4) loci, which we chose for their high expression. Strong sequence conservation prevented the investigation of several other loci.

Promoters of olfactory receptor genes. In this example of a region of chromosome 7, which is symbolized as the horizontal axis, the transcription start sites have been plotted as vertical bars proportional to the number of nanoCAGE tags aligned, normalized in tags per million (TPM). Activity is displayed upward for the forward strand and downward for the reverse strand. (Blue boxes) The positions of the OR transcript models, with their names following the Olfr convention. Note that the transcript models contain only the predicted coding sequence of the ORs and lack the 5′ and 3′ UTRs. Sequence conservation and expression filtering identified the three largest peaks as promoters of their downstream OR gene model. (Yellow) The revised gene models.
Predicted transcriptional control of OR genes
We next used the 955 OR promoters to identify cis-regulatory elements and TFs that may control OR gene transcription. We analyzed the TFBSs contained in genomic sequence in the regions from −300 (upstream) to +100 (downstream) of the major TSS. This interval maximizes the signal/background ratio in identifying TFBSs (Suzuki et al. 2009). We compared these OR promoters with two reference sets of promoters for ∼3000 non-OR genes that are expressed in the MOE or in the FANTOM3 CAGE libraries and that present the same general class of sharp promoters without CpG islands (Carninci et al. 2006). To detect over-representation of known TF binding site motifs, we used our in-house statistical software tools along with positional weight matrices (PWMs) from the JASPAR (Portales-Casamar et al. 2010) and TRANSFAC Public (Matys et al. 2003) databases, which altogether identify TF binding motifs. We also generated a PWM for EBF1 from 26 experimentally validated binding sites collected from the literature (Supplemental Table S3). Our results reveal in OR promoters an over-representation of TF binding site motifs that are bound by homeodomain proteins (NKX family, FOX family), SOX family proteins, EBF proteins, the MADS-box protein MEF2A, and the TATA-binding protein TBP (Fig. 3; Table 1: Supplemental Table S4; results are summarized in Fig. 3A).
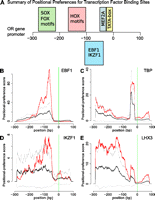
Transcription factor binding profile over OR promoters. (A) Summary of positional preferences for TFBS in relation to the major TSS. (B–E) TF binding site motif profiles in OR (red) versus reference set of promoters (black). (Gray lines) 99.99% confidence intervals around the mean TFBS positional preference profile for the background set used. (Vertical green lines) The position of the major TSS.
TFBS analysis of the MOE
For a de novo discovery of motifs, we used MEME (http://meme.sdsc.edu/) and Amadeus (http://acgt.cs.tau.ac.il/amadeus/) with a general set of sharp, non-CpG promoters as reference. We then compared the motifs with JASPAR and TRANSFAC public databases using STAMP (http://www.benoslab.pitt.edu/stamp/). While MEME uncovers only the EBF1 binding site with high confidence, Amadeus predicts additional motifs similar to those for HOX and FOX. The over-representation of FOX, HOX, and EBF1 TF binding site motifs in OR gene promoters is thus confirmed by independent computational approaches.
By exploring the PWMs according to the fold increase of their frequency in OR promoters compared with reference sets, we noticed a strong enrichment in OR promoters for the PWM of POU (OCT) (94%–18% vs. 30%–2%), MEF2A (62%–20% vs. 14%–4%), and TBP (85% vs. 30%) (Table 1). NanoCAGE data confirm the expression of Pou2f1, Pou6f1, Mef2a, and Tbp in the MOE (Table 1). We also found binding motifs for TFs whose related gene expression as tags in the nanoCAGE libraries was not detected (Supplemental Table S4); among these cases, the LHX3 TF binding site motif is present in as many as 88% of the OR promoters and only in 15% of the FANTOM3 reference promoter set. Although we could not detect Lhx3 expression in the MOE, we confirm the expression of Lhx2, a related gene. LHX2 and LHX3 have been shown to bind the same probe by electromobility shift assay (Roberson et al. 1994; Bach et al. 1995), and LHX2 is essential for OSNs identity (Hirota and Mombaerts 2004; Kolterud et al. 2004; Hirota et al. 2007).
Sharp promoters without CpG islands, which we have here shown to prevail in OR genes, are often bound by TFs within a constrained spacing range relative to the TSS (Roider et al. 2009). To determine the spatial preferences of the over-represented TF binding site motifs, we plotted the distribution of the corresponding TF binding site motifs in the region relative to the major TSS (Fig. 3B–E). Several of the over-represented TF binding site motifs exhibit a strong spatial preference (Table 1), the strongest signal coming from EBF1 (Fig. 3B) between 50 and 150 bp upstream of the major TSS, followed by the very precise TATA-box (TBP) signal at the expected spacing of −33 to −29 (Fig. 3C), and the enrichment of homeobox core TF binding site motifs peaking at 100–150 bp upstream of the major TSS (Fig. 3A). This distribution confirms the previous identification of putative HD and EBF binding sites in the promoters of several OR genes (Vassalli et al. 2002). For TBP (Fig. 3C) and MEF2A, we identified additional potential binding sites downstream from the major TSS. An enrichment of the PWMs of IKZF1 (also known as IKAROS) was also detected (Fig. 3D).
Class I and Class II OR genes belong to distinct phylogenetic categories, and it has been previously suggested that their transcription may be controlled by differing regulatory elements (Hirota et al. 2007); we therefore carried out a systematic comparison of promoters for Class I versus II OR genes. The promoter and coding sequence of Class I receptors show a tendency to be more conserved (p < 0.05, one-tailed Wilcoxon test). Class II OR promoters have an increased frequency of HOX and FOX TF binding site motifs with special preference for LHX3 (p = 9.9 × 10−4, Fisher's exact test, two-tailed). They are also more frequent in the conserved noncoding elements between the OR genes (p = 4.0 × 10−5). We found a similar over-representation in Class II OR promoters for a LHX2 PWM obtained from the UniProbe database, and a binding sequence consensus (ymATTAnnTAATkr) derived from the weblogo (Hu et al. 2009) after dimerization (Roberson et al. 1994) (p = 1.0 × 10−4). We did not find over-represented TFBS for Class I OR promoters.
AACTTTTTAATGA is a sequence conserved within the H element and the P sequence, two regulatory regions that control OR genes' expression (Fuss et al. 2007; Bozza et al. 2009). To assess the potential involvement of this particular sequence in OR genes regulation, we examined its conservation in OR promoters allowing up to four mismatches, and we found it to be over-represented around −150 bp with respect to the major TSS, a positional preference similar to HOX TF binding site motifs.
It has been speculated that locus control regions or long-range regulatory elements have a general role in regulating OR gene expression (Lane et al. 2002). Consequently, we assessed the presence of conserved regions in noncoding elements found up to 50 kb from OR transcript 5′ ends. We found that noncoding elements around Class I genes have deeper conservation than around Class II genes (p < 2.2 × 10−16, one-tailed Wilcoxon test), reflecting perhaps their grouping in a single cluster on chromosome 7. In addition, we noticed that Class I genes are more likely than Class II genes to have a conserved noncoding element nearby (92% vs. 84%, respectively; p = 0.020, one-tailed Fisher's exact test). Comparison of TFBSs content around these elements in Class I and II OR genes shows over-representation of HOX TF binding site motifs in conserved elements at Class II loci, consistent with promoter analysis.
In vivo interaction of EBF, TBP, and MEF2A TFs with OR promoters
We assayed the physical association of three TFs to promoter regions of OR genes in vivo by chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR). The OR genes were selected for their high expression levels according to nanoCAGE data. EBF-binding motifs are strongly enriched in our analysis; by using an anti-pan EBF antibody, we confirmed EBF immunoreactivity in the nucleus of OSNs (Fig. 4A). Chromatin from MOE of mice at postnatal days 22–30 was purified and immunoprecipitated with specific or control (IgG) antibodies. We quantified bound genomic DNA by qPCR using primers for the promoter regions of Olfr110 (MOR249-2) and Olfr794 (MOR114-11), both containing a putative EBF binding site. Figure 4B shows that EBF binds in vivo the predicted OR promoter regions. We also tested two additional TFs that have not been implicated previously in the regulation of OR gene expression, TBP and MEF2A, which were selected for their high expression level according to nanoCAGE data and for the availability of specific antibodies. We confirmed their nuclear localization in the OSNs by immunohistochemistry (Fig. 4A). TBP is expressed across all layers of the MOE, while MEF2A expression is more restricted to cells in basal layer. We confirmed by ChIP-qPCR the physical association of TBP and MEF2A with chromatin regions upstream of Olfr279 (MOR122-1), Olfr683 (MOR40-1), and Olfr1106 (MOR172-6) (Fig. 4B). We used sequences from the Hist2h promoter as a positive control for TBP ChIP. These data strongly suggest that EBF, TBP, and MEF2 are associated with assayed OR gene promoters in the mouse MOE.
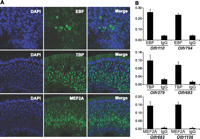
EBF, TBP, and MEF2A proteins are expressed in the MOE and physically associate with the promoters of Olfr110, Olfr279, Olfr683, Olfr794, or Olfr1106 in vivo. (A) Immunohistochemistry of MOE from mice at postnatal day 21, with anti-EBF, anti-TBP, and anti-MEF2A antibodies (green). Nuclei were visualized with DAPI (blue). Scale bars, 30 μm (top row), 60 μm (center and bottom row). (B) ChIP assays were performed on MOE from mice at 22–30 d postnatally. Chromatin was immunoprecipitated with anti-EBF, anti-TBP, and anti-MEF2A antibodies. Normal rabbit IgGs were used as negative control. Enrichment of promoter sequences in the immunoprecipitates was determined by qPCR, using specific primers. The relative abundance of target DNA was expressed as percentage of total input chromatin. Data were obtained from two independent experiments. Statistical analysis: p < 0.05 in all cases.
Validation of an OR promoter in transgenic mice
To test if sequences defining an OR promoter identified by nanoCAGE are sufficient for its expression, we generated transgenic mice expressing a LacZ reporter under the control of sequences flanking the major TSS of an OR gene (Vassalli et al. 2011), which did not include any OR coding sequence (Serizawa et al. 2003). We used a genomic fragment from the Olfr160 (M72) locus that consists of 143 bp upstream of the Olfr160 major TSS and the first 154 bp of the noncoding exon of Olfr160, and named this construct Tg-M72(V4)-LacZpA (Fig. 5A–C; Vassalli et al. 2011). This 298-bp segment contains a sharp promoter (Fig. 5A) and multiple EBF1 and MEF2A binding sites both upstream of and downstream from the TSS as defined by nanoCAGE (Fig. 5B). Nine out of 12 transgenic lines and 5/5 transgenic founders showed expression in the turbinates. An X-gal-stained whole mount of a mouse from line 7 is shown in Figure 5C. Reporter expression is somewhat ventralized relative to the normal pattern of Olfr160 transcript localization. Thus, a 298-bp sequence centered on the major TSS of Olfr160 and containing binding sites for EBF and MEF2A is able to generate promoter activity in the OSNs of transgenic mice.
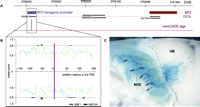
Transgenic reporter assay of the promoter activity of sequences flanking a nanoCAGE TSS. (A) Schematic depiction of the locus of Olfr160 (M72) on mouse chromosome 9. (Red) OLFR160 coding sequence; (blue) Olfr160 transgenic promoter; (purple) nanoCAGE tags; (black) ESTs. (B) Position weight matrix (PWM) scanning of the promoter sequences with matrices for EBF1 (green) and MEF2A (blue). Highlighted are the regions where the signal is above a 0.8 score threshold. (C) Whole mount of a mouse from transgenic line 7 shows X-gal-labeled cells predominantly in the middle domain of the MOE (black arrows). Labeled axons spread over a large domain in the middle aspect of the medial half-olfactory bulb, a probable consequence of the absence of an intact OR CDS in the transgene. The dotted gray line follows the shape of the cribiform plate separating the MOE from the olfactory bulb (OB).
Discussion
With the combination of LCM and nanoCAGE, we have provided a comprehensive identification of TSSs and promoters in the mouse MOE. The use of LCM allowed us to harvest the target tissue minimizing contamination by neighboring non-olfactory tissues. The newly developed nanoCAGE technology was instrumental to describe the 5′ ends of transcripts from small, fixed, ex vivo samples for both poly(A)+ and poly(A)− transcripts. Deep sequencing enabled us to collect information on expressed promoters at unprecedented depth, as confirmed by the detection of OR genes that are expressed in <1% of cells. We have thus determined the promoter architecture of 87.5% of the mouse OR genes, and we have provided a thorough characterization of the transcriptional landscape of OR loci.
The architecture of OR promoters
We have associated TSSs to 955 mouse OR genes, thereby defining a comprehensive picture of their promoter map at a single-base resolution. In contrast with the archetype of >75% of mammalian promoters, OR genes have sharp promoters exhibiting a dominant TSS. One of the most relevant features of sharp promoters is the spatially constrained distribution of cis-regulatory elements. They often contain a well-defined TATA-box (Ponjavic et al. 2006) but no CpG island. In contrast, ∼90% of promoters that overlap a CpG island lack a TATA-box. Exceptions to this general rule are tissue-restricted transcripts in the brain that tend to be controlled by CpG island-overlapping, broad promoters (Gustincich et al. 2006). Broad promoters control genes that are typically expressed more widely, including nearly all so-called housekeeping genes (Carninci et al. 2006). In contrast with some other works (Glusman et al. 2000; Sosinsky et al. 2000; Clowney et al. 2011) and in line with the comparative analysis of Bulger et al. (2000), we found a well-defined TATA-box in 21% of both Class I and Class II OR genes at a higher frequency if compared with other sharp promoters. Most of the other OR genes display a weaker TATA-like motif at the expected position. To exclude that the difference for the detection of the TATA-box stems from the bioinformatics rather than from molecular biology, we searched in Clowney et al.'s OR promoters for TBP, LHX3, EBF1, and IKZF1 PWMs following our approach and confirmed the absence of a clear TBP signal in their data (Supplemental Fig. S3). This could be the consequence of sequence biases caused by the use of hybridization in the tiling arrays method, or more generally its lack of single-nucleotide resolution. OR promoters have therefore characteristics of non–nervous tissue–restricted genes. In the brain, the list of tissue-restricted genes that have TATA-box, non-CpG island sharp promoters includes retina-specific genes such as opsins, retbindin, and retinal S-antigen. Resemblances in the transcriptional control of those genes may be due to a similar role as sensory transduction elements with an early origin in evolutionary history, thus sharing the more ancient type of tissue-restricted regulation that is based on sharp, TATA-boxed promoters.
Transcriptional regulation of OR expression
Our large-scale approach offers the opportunity to predict the TFs that are involved in the transcription of OR genes in the MOE. We identified EBF1 and IKZF1/IKAROS TF binding site motifs in the EBF binding site region, as well as HOX and FOX TF binding site motifs in the HD binding site. A new binding site region was defined close to the major TSS, containing a TF binding site motif for the MADS-box protein MEF2A, identifying a new potential control site for OR gene regulation.
The identification of EBF1 confirms the validity of our approach: EBF1 has been the first TF to be implicated in the regulation of the OSN-specific genes G(olf) (Gnal), adenylyl cyclase III (Adcy3), the olfactory-specific subunit A2 of the cyclic nucleotide-gated ion channel (Cnga2), and Gγ8 (Gng8) (Travis et al. 1993; Wang et al. 1993, 1997). The DNA binding site for EBF1 shares its consensus sequence with two other family members, EBF2 and EBF3, and it shows a strong spatial preference for positioning between 50 and 150 bp upstream of the major TSS, which also occurs in the reference set of 3000 promoters for non-OR genes in the MOE. Such a widespread and precise positioning in all sharp, CpG-less promoters argues for a more general function of the EBF1, EBF2, and EBF3 proteins in regulating the transcription of adult tissue–restricted genes than in the control of OR gene expression per se. Putative binding sites for the zinc finger transcription factor IKZF1/IKAROS are known to be conserved between the mouse and human P3 and P4 promoter regions (Lane et al. 2001). We observed that for EBF1 and IKZF1/IKAROS, the distance between TF binding site motif peaks and the major TSSs of OR genes is very similar despite their unrelated binding profiles (Fig. 3D). Interestingly, EBF1 has also a prominent function in defining the transcriptional profile of lymphocyte progenitors (Nutt and Kee 2007).
The association of HOX and FOX TF binding site motifs has been strengthened by the strict conservation of distances between their binding sites and the major TSSs of the OR genes they control. Interestingly, their mapping coincides with the well-characterized HD binding site in the OR promoter region. Among them, PWMs of POU (OCT) genes were strongly enriched (Table 1; Supplemental Table S4). Importantly, our nanoCAGE libraries provided evidence for expression of transcripts corresponding to enriched TFBS, including Pou6f1, Foxa2, and Foxg1. Using the in situ hybridization expression pattern database Eurexpress (http://www.eurexpress.org), we confirmed that their expression is not restricted to the sustentacular cells that were included in our microdissection (Supplemental Fig. S1). Due to the similarity of their binding profiles, it is difficult to ascertain which Fox gene product binds OR promoters. Two possible candidates are FOXA2, which has been detected in both adult (Besnard et al. 2004) and embryonic (http://www.eurexpress.org) MOE, and FOXG1, which is essential for the production of mature OSNs in a cell-autonomous fashion (Duggan et al. 2008).
We also found TF binding site motifs for TFs that do not seem to be expressed in the MOE (Supplemental Table S4), such as LHX3, which shows strong prevalence in OR promoters; we hypothesize that this motif can be bound by the related protein LHX2, which can be expected to have a similar binding site. This hypothesis is corroborated by the presence of an LHX3 binding site motif in 54% of the MOE reference promoter set, but only 15% of the FANTOM3 reference set. Furthermore, LHX3 TF binding site motifs are over-represented in the promoters of Class II OR genes, providing a molecular basis for the selective loss of this receptor class in Lhx2 knockout mice (Hirota et al. 2007).
We also show that approximately half of the expressed OR loci contain antisense transcripts, with the majority of them located within the coding regions. It will be interesting to test whether antisense and other noncoding RNAs originate in chromatin regions that are involved in yet-unknown three-dimensional structures that may play a role in the monoallelic and monogenic expression of OR genes. Indeed, molecular players of chromatin assembly and remodeling have been shown to be enriched in OSNs (Sammeta et al. 2007). An additional highly represented class of promoters or at least capped 5′ ends of RNAs is located in the 3′ UTR of OR genes. It is still unclear whether they drive the expression of small noncoding RNAs or of unknown transcripts that may further contribute to the transcriptional regulation of OR genes, for example, acting as a sponge to compete for microRNA target sites (Tay et al. 2011).
In vivo activity of an OR promoter transgene
Experimental analysis of OR gene regulation has been confined to a few OR genes, by applying transgenic mouse technology to identify fragments of OR loci that reproduce some or all of the features of OR gene expression. Transgenes of ∼9 kb for Olfr151 and Olfr16 (Vassalli et al. 2002; Rothman et al. 2005) and 10.5 kb for Olfr157 (MOR262-12) (Zhang et al. 2007) are the smallest genomic fragments (minigenes) that closely reproduce the features of endogenous OR genes. For the Olfr151 minigene, a 161-bp region upstream of the major TSS is required for transgene expression (Rothman et al. 2005). Olfr16 transgene expression is dependent on a 405-bp region upstream of the TSS, which consists of a 257-bp fragment of a LINE-1 followed by 148 bp of unique sequence. Conserved sequence TF binding site motifs were indeed identified in the regions upstream of the TSS including a putative HD and an EBF-binding site. By site-directed mutagenesis, these TF binding site motifs were then shown to be important in vivo for OR gene expression (Rothman et al. 2005). Conversely, analysis of Ebf2 and Ebf3 knockout mice revealed marked defects in the projection of OSN axons to the dorsal part of the olfactory bulb (Wang et al. 2004). Mice with a knockout mutation in the homeobox-containing gene Emx2 show defects in OSN maturation and altered OE structure (McIntyre et al. 2008). Furthermore, LHX2 interacts with a HD binding site in the Olfr16 promoter and is required for expression of Class II OR genes but not for Class I OR genes and/or the maturation of the corresponding OSNs (Hirota and Mombaerts 2004; Kolterud et al. 2004; Hirota et al. 2007).
Here we show that mosaic, OSN-specific expression can be conveyed to the taulacZ reporter by a very small genomic segment from the Olfr160 locus flanking the major TSS and containing the binding sites that we have identified. The taulacZ reporter itself is completely inactive in the absence of a functional promoter (Rothman et al. 2005). Unlike most other published OR transgenes such as the minigenes (Vassalli et al. 2002; Rothman et al. 2005; Zhang et al. 2007), this transgene does not contain an OR coding sequence. Accordingly, stained axons do not coalesce into a few glomeruli but project diffusely over the olfactory bulb, presumably reflecting the expression of a wide variety of endogenous ORs by the transgene-expressing OSNs (Vassalli et al. 2011). Within the 298 bp of the Tg-M72(V4)-LacZpA promoter transgene, one HD binding site and several EBF and MEF2A binding sites are present just upstream of the major TSS, suggesting that these sequences are sufficient for OSN-specific expression of the reporter gene. Although the Olfr160 promoter transgene resembles the endogenous spatial expression pattern, expression extends to the ventral MOE. Such deviations from the endogenous expression pattern have also been reported with larger transgenes (Vassalli et al. 2002; Rothman et al. 2005), suggesting that the full control region may contain additional sequences that have a negative regulatory role across the MOE.
The widespread resemblance between all OR gene promoters, their ability to drive broad expression in the MOE with minimal sequences around the major TSS, and the absence of TF binding site motifs specific for spatial or phylogenetic subsets of OR genes suggest that the other key parts responsible for monoallelic and monogenic expression mechanisms are to be searched for outside the core promoter. Cis-acting enhancers like the H element (Serizawa et al. 2003) are natural candidates. Indeed, our TF binding site motif analysis of the evolutionarily conserved regions upstream of the OR gene promoters underlined HD TF binding site motifs similar to those of the H element. However, we did not find features in the upstream conserved elements that correlate with spatial or phylogenetic subsets of OR genes and that would have suggested a determinant role for such enhancers in the choice of a single olfactory receptor. Accordingly, deletion of the H element only affects its local OR gene cluster (Fuss et al. 2007).
Among the other mechanisms that can globally control gene expression, epigenetic modifications of the DNA or the chromatin may be a good complement, for instance, to select a single locus in which interaction between a shared enhancer and the stereotyped OR core promoters would then trigger the selection of a single OR gene. Interestingly, nanoCAGE tags strongly indicate the presence of noncoding transcripts, which are observed in other epigenetic systems of monoallelic expression (Yang and Kuroda 2007). Further experiments are needed to investigate the role of these novel promoters and transcripts. In particular, it will be very important to analyze them in the context of single cells, where it can be seen if they are active in the same locus as the selected OR, or in a different locus or OR cluster.
Methods
Preparation of samples for LCM
C57BL/6J mice were sacrificed by intraperitoneal injection of pentobarbital, 300 mg/kg body weight. After decapitation, the skin and the jaw were removed from the heads, and the samples were left overnight in 1× ZincFix fixative (BD Biosciences) diluted in DEPC-treated water. After a 4-h cryoprotection step in a 30% sucrose/1× ZincFix solution, heads were included in Frozen section medium Neg-50 (Richard Allan Scientific) and left for 15 min on dry ice. Frozen blocks were brought into a cryostat (Microm International) and left for 60 min at −21°C. Serial coronal sections of mouse heads (16 μm) were cut with a clean blade, transferred on PEN-coated P.A.L.M. MembraneSlides (P.A.L.M. Microlaser Technologies), and immediately stored at −80°C. Before usage, the slides were brought to room temperature and air-dried for 2 min. For 100 individual serial coronal sections in total encompassing all of its four zones, the MOE was morphologically identified, marked, microdissected, and catapulted with a Zeiss P.A.L.M. LCM microscope (Carl Zeiss Inc.) in P.A.L.M. tubes with adhesive caps. After the harvest, 10 μL of lysis buffer (Stratagene) was added in each cap; the samples were left for 10 min at room temperature, centrifuged at 6000g for 10 min, and stored at −80°C. RNA from the pooled samples was extracted and purified with the Absolutely RNA Microprep Kit (Stratagene), and 2.4 μg was recovered.
nanoCAGE
nanoCAGE libraries were prepared as described (Plessy et al. 2010). In brief, total RNA was concentrated by centrifugal evaporation at room temperature in the presence of trehalose, sorbitol, the template switching (TS) oligonucleotides, and the random (N15) or polythymine (dT18) reverse-transcription primers. All oligonucleotides are listed in Supplemental Table S5 with their sequence. cDNA was synthesized with SuperScript II and amplified by semi-suppressive PCR14 with Ex Taq (TaKaRa) with the forward (FSS) and reverse (RSS) primers, following the program 5 min at 95°C, n × (10 sec at 95°C, 15 sec at 65°C, 2 min at 68°C), where n was determined according to the concentration of each cDNA preparation. Tags were cleaved with EcoP15I, purified through the Microcon YM-100 membranes (Millipore), and then concentrated on Microcon YM-10. Bar codes were introduced by ligation with a DNA duplex adaptor made by annealing two oligonucleotides (L1 and L2, where the nucleotides labeled as NN are the bar code) (Supplemental Table S5). The resulting tags were amplified by PCR with Ex Taq (TaKaRa) using the FT and RT primers, with the program 5 min at 95°C, n × (10 sec at 95°C, 10 sec at 68°C), where n was determined according the concentration of each ligation reaction, and purified on an 8% polyacrylamide gel. Samples were sequenced using the Illumina GA sequencer. The read length was 36 bases, and the average tag length was 25 bases after extraction (we observed small fluctuations in the distance between cleavage site and binding site for EcoP15I).
Tags were mapped to mouse genome version mm9 (NCBI build 37) using the Vmatch program (http://www.vmatch.de), with a minimum match length of 21 bases and a maximum of one error. Tags mapping the mouse ribosomal DNA sequence were eliminated. The “best” match for each tag was then calculated as the alignment with the highest Vmatch score. Tags mapping to more than 10 genomic loci at the best match level were then removed. MuMRescueLite (http://genome.gsc.riken.jp/osc/english/dataresource/) was then run on each library separately with a window size of 200 bp, producing weighted values for multi-mapping tags occurring within 100 bp of a single map tag on the genome.
Preparation of mouse OE RNA for 5′- and 3′-RACE validation and RT-PCR
Adult wild-type C57BL/6J mice were sacrificed by carbon dioxide inhalation. The MOE was dissected from the heads and rapidly snap-frozen in liquid nitrogen. Total RNA was extracted with TRIzol reagent (Invitrogen). The RNA sample was treated with DNase (Ambion) for 1 h at 37°C, cleaned with an RNAeasy Kit (QIAGEN), and aliquoted in RNase free LowBind tubes (Eppendorf). 5′-RACE-ready cDNA was synthesized with a GeneRacer Kit (Invitrogen).
3′-Deep-RACE PCR was performed using the Illumina Genome Analyzer II. In brief, 1 μg of total RNA was reverse-transcribed as for a nanoCAGE library, except for the reverse-transcription primer RRT, and the cDNAs were amplified by semi-suppressive PCR with the FSS and RR primers, to ensure that only capped molecules were analyzed. Nested RACE-PCR was then conduced with gene-specific forward primers designed with scripts available upon request, and 3′ RACE primers; for the outer nested PCR, the ORP reverse prime and gene-specific forward primers. The inner PCR was done with the IRP reverse primer and gene-specific primers, in which CAAGCAGAAGACGGCATACGA is a tail to accommodate sequencing on the Genome Analyzer II (Illumina) with the DS primer.
For RT-PCR, first-strand cDNA was synthesized using the SuperScript II enzyme (Invitrogen) in 50-μΛ reactions for 2 h at 42°C. PCR was performed by adding 1 μL of first-strand reaction to a mix containing 5 U of Ex Taq DNA polymerase, 10× buffer, dNTPs mix 2.5 mM each (all reagents from TaKaRa, Japan), 50 pmol forward and reverse primers, and nuclease-free water to a final volume of 50 μL. Forward exon-spanning primers were used to avoid unwanted amplification of residual genomic DNA.
Mapping tag clusters to OR genes
We extracted conserved blocks from the MULTIZ alignment track in the UCSC Mouse Genome Browser (mm9). We clustered conserved blocks of mouse–rat, mouse–human, and mouse–dog together, if they were at most 4000 bp apart. These conserved block clusters were attributed to RefSeq OR transcript models. For each OR, we selected the conserved block clusters between 100 bp downstream from the model's 5′ end (corresponding in most cases to the start of the coding sequence), and the 3′ end of the directly upstream RefSeq on the same strand, extended by 500 bp in order to make sure that transcription arising from within the upstream gene's 3′ UTR (Carninci et al. 2006) would not be falsely included. We also limited the span of the upstream region to 15 kb in case the conserved block cluster was longer and no upstream RefSeq transcripts were to constrain its boundary. We only accounted for regions within those boundaries that did not correspond to repeats obtained from the RepeatMasker track in the UCSC Mouse Genome Browser (mm9). We then associated the tag cluster with the highest expression level within this region to the corresponding downstream OR gene.
Detection of conserved regions and elements
For the identification of conserved noncoding regions in the neighborhood of OR genes, we used Phastcons conserved elements for 20 placental mammals with the mouse mm9 assembly as reference, available from the UCSC Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables) as the table “Mammal El (phastConsElements30wayPlacental)” in the “Conservation track” of the “Comparative Genomics” group. We have retained Phastcons conserved elements if they do not overlap with any of the following: mouse ESTs, mRNA exons, RefSeq exons, exons of other species genes mapping to the mouse genome, and CAGE tag clusters. We used the same algorithm for the analysis on CNEs, with as a background 3000 randomly chosen CNEs that are not nearby OR genes. Only CNEs larger than 10 bp were considered.
Over-representation of repeat element family in evolutionarily conserved clusters of OR genes
Repeat elements mapping to the mm9 genome were downloaded from UCSC, and summed expression of members of all repeat families located on or outside evolutionarily conserved clusters of OR genes was gathered. One-tailed Fisher tests were used to calculate the potential overexpression of specific repeat families in CLiC compared with that on the entire genome. Specifically, the 2 × 2 contingency table built to accept or reject the hypothesis that specific repeat families were over-represented in CliCs was composed of (1:1) count of tags overlapping of repeat of family X in CLiCs, (1:2) count of tags overlapping of repeat of family X not in CLiCs, (2:1) count of tags overlapping any nonfamily X repeats in CLiCs, and (2:2) count of tags overlapping any nonfamily X repeats not in CLiCs.
TFBS over-representation analysis
We compared the normalized total number of TFBS profile hits in OR gene promoters to those of our reference set and obtained significantly over-represented TFBSs in OR gene promoters. The significance of the obtained over-represented TFBS was assessed by performing a similar computation on 10,000 independent random resamplings of promoters among all promoters, OR promoters, and non-OR sharp promoters, and counting the number of sets with equal or higher value to the original total hit count divided by 10,000, thus obtaining an estimate of the P-value for each TF binding site motif:OR promoter association. We also assessed the significance of the number of sequences in the target promoter set having a given TF binding site motif using random sampling with replacement, counting the number of random sets that had a higher or equal number of sequences containing that TFBS hit, and calculated the P-value by dividing it by the total number of random sets. We retained as significantly over-represented, OR promoter TF binding site motif associations for which both P-values were <0.005.
TFBS positional-preference analysis
We computed positional-preference profiles for OR gene promoters. We computed similar profiles for 1000 sets, of a size similar to the set of OR promoters, randomly sampled from the union of all OR promoters and 3000 sharp, CpG-less promoters. Assuming that for each position positional-preference scores are distributed normally, we calculated the mean and 99.99% confidence interval values. Comparing them to scores obtained with OR promoters using Z-score, we calculated the P-value associated with the over-representation of a TF binding site motif at each position.
We finally ranked all of the TFBS positional-preference profiles according to their lowest P-value, requiring that the profile local minima not overlap with a TATA-box region since genuine TATA motifs can be confounded with AT-rich parts of some TFBS, and selected the top 30 profiles.
Chromatin immunoprecipitation (ChIP)
The MOE was dissected from C57BL6/J mice at postnatal day 22–30. Six to seven mice were used for the preparation of chromatin. Tissues were minced on ice in 10 mL of DMEM. For cross-linking, formaldehyde was added directly to the culture medium to a final concentration of 1% and incubated for 15 min at room temperature. The reaction was stopped with 0.125 M glycine for 5 min. Tissues were collected by a brief spin, washed with PBS, and homogenized in PBS supplemented with protease inhibitors (Roche) using a Dounce homogenizer with a loose pestle. Solitary cells were filtered through a cell strainer (BD Falcon) and collected by spinning at 2000g for 5 min. The cell pellet was resuspended in 50 mL of LB1 buffer supplemented with protease inhibitors (50 mM HEPES-KOH at pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) and incubated for 10 min at 4°C with rocking. Cells were then collected by centrifugation at 1350g for 5 min and incubated with 5 mL of LB2 buffer with protease inhibitors (10 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) for an additional 10 min. Isolated nuclei were recovered by centrifuge at 1350g for 5 min at 4°C and then resuspended in 2.5 mL of immunoprecipitation buffer LB3 plus anti-protease cocktail (10 mM Tris-HCl at pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.5% Na-deoxycholate, 0.5% N-lauroylsarcosine). About 200 mg glass beads (Sigma, G-1277) were added, and nuclei were sonicated on ice (12 cycles at 50% maximum power for 30 sec each plus 30 sec on ice; Soniprep 150 MSE, Sanyo). Fragmented chromatin (genomic fragments with a bulk size of 200–1000 bp) was separated from intact nuclei and cellular debris by centrifugation at 20,000g for 15 min at 4°C. An aliquot of lysate (500 μL) was then diluted in LB3 buffer supplemented with 1% Triton X-100 and protease inhibitors to a final volume of 2 mL per IP (equivalent to 35–50 μg of DNA from ∼60 mg of tissue). Chromatin immunoprecipitation was performed with magnetic beads (Dynabeads; Invitrogen) following the protocol as described (Schmidt et al. 2009). For each ChIP, 2 μg of antibody was used. Anti-EBF (sc-33552), anti-TBP (sc-204X), and anti-MEF2A (sc-313X) were from Santa Cruz. ChIP-grade control rabbit IgG was from Cell Signaling (#2729).
qPCR was performed using SYBR Green PCR Master Mix and an iCycler IQ Real-Time PCR System (Bio-Rad). Enrichment of chromatin binding was calculated relative to total input, as described previously (Frank et al. 2001).
Transgenic mice
The transgenic construct Tg- M72(V4)-LacZpA (Vassalli et al. 2011) is publicly available as plasmid 15607 from Addgene. Founders were bred with C57BL/6J mice to transmit the transgene through the germline. Line 7 has been cryopreserved as sperm at The Jackson Laboratory (Bar Harbor, Maine, USA), and is publicly available as stock number 7974, official strain name B6;CBA-Tg(Olfr160-taulacZ)V4-7Mom/MomJ.
Data access
nanoCAGE and Deep-RACE sequences have been submitted to the DNA DataBank of Japan Sequence Read Archive (DRA) under accession numbers SRP000696 and DRA000474. A bibliographic survey of EBF1 binding sites was deposited in the JASPAR database, accession number MA0154.1.
Acknowledgments
We are indebted to Valerio Orlando (Telethon Research Institute, Rome, Italy) for his help with the ChIP experiment and to all the members of the Stefano Gustincich laboratory. We thank Mylène Josserand for help with the Deep-RACE experiments, and Anna Menini (International School for Advanced Studies, Italy) for helpful discussions. This work was funded by a grant of the 6th Framework of the European Union commission to the NFG consortium (P.C. and S.G.); a Grant-in-Aid for Scientific Research (A) No. 20241047 to P.C.; a Research Grant for RIKEN Omics Science Center from the Japanese Ministry of Education, Culture, Sports, Science and Technology to Y.H.; U.S. National Institutes of Health grants to A.V. and P.M.; and a Next Generation World-Leading Researchers (NEXT Program) grant from the Japanese Society for the Promotion of Science to P.C. C.P. was supported by the Japanese Society for the Promotion of Science long-term fellowship number P05880. S.G. was funded by a career developmental award from the Giovanni Armenise–Harvard Foundation.
Authors' contributions: C.P., G.P., C.C., A.V., D.L., C.V., R.S., and S.Z. performed the experiments; C.P., J.K., C.O.D., Y.H., and P.C. were involved in the large-scale data production; C.P., N.B., A.A., J.S., G.J.F., and B.L. analyzed the data; and C.P., P.M., S.G., and P.C. wrote the manuscript.
Footnotes
-
↵15 Corresponding authors.
E-mail Boris.Lenhard{at}bccs.uib.no.
E-mail gustinci{at}sissa.it.
E-mail carninci{at}riken.jp.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.126201.111.
- Received May 12, 2011.
- Accepted November 30, 2011.
- Copyright © 2012 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








