
Copy-number-aware differential analysis of quantitative DNA sequencing data
- Mark D. Robinson1,2,3,7,
- Dario Strbenac3,
- Clare Stirzaker3,6,
- Aaron L. Statham3,
- Jenny Song3,
- Terence P. Speed4,5 and
- Susan J. Clark3,6
- 1Institute of Molecular Life Sciences,
- 2SIB Swiss Institute of Bioinformatics, University of Zurich, CH-8057 Zurich, Switzerland;
- 3Epigenetics Laboratory, Cancer Research Program, Garvan Institute of Medical Research, Sydney 2010, New South Wales, Australia;
- 4Bioinformatics Division, Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne 3052, Victoria, Australia;
- 5Department of Medical Biology, University of Melbourne, 3050, Victoria, Australia;
- 6St. Vincent's Clinical School, University of New South Wales, Sydney 2010, New South Wales, Australia
Abstract
Developments in microarray and high-throughput sequencing (HTS) technologies have resulted in a rapid expansion of research into epigenomic changes that occur in normal development and in the progression of disease, such as cancer. Not surprisingly, copy number variation (CNV) has a direct effect on HTS read densities and can therefore bias differential detection results. We have developed a flexible approach called ABCD-DNA (affinity-based copy-number-aware differential quantitative DNA sequencing analyses) that integrates CNV and other systematic factors directly into the differential enrichment engine.
All normal cells carry the same DNA sequence, yet distinct cell types result from gene expression patterns that are controlled by a combination of genetic and epigenetic mechanisms. In cancer, genetic and epigenetic changes result in altered gene expression patterns, such as up-regulation of oncogenes and down-regulation of tumor-suppressor genes (Jones and Baylin 2007; Stratton 2011). Specifically, mutations in the DNA sequence or changes in copy number can alter how these genes are regulated or expressed, as can nonsequence epigenetic features such as chemical (e.g., DNA methylation or histone modifications) or structural makeup (e.g., nucleosome occupancy). Advances in microarray and especially high-throughput sequencing (HTS) technologies have driven a deeper exploration of genetic and epigenetic phenomena, resulting in several large data collection projects (Jones et al. 2008; Bernstein et al. 2010; International Cancer Genome Consortium 2010; Stratton 2011) as well as many smaller scale studies. Statistical and computational tools for processing and interpreting these data sets are maturing, and altogether these give exciting prospects for the understanding, detection, prevention, and treatment of cancer and other diseases.
Recently, we highlighted that comparisons between cancer and normal epigenomes need to be informed by genomic changes (Robinson et al. 2010b,c). Specifically, copy number variation (CNV) has a direct effect on read densities of affinity (or enrichment)-based assays (e.g., chromatin immunoprecipitation [ChIP] and methylated DNA capture [MBDCap]); we refer to these techniques collectively as qDNA-seq, since they all provide a quantitative epigenetic readout at a specific loci. In these assays, a subset of target DNA fragments are captured, prepared, sequenced, and mapped to a reference genome. Enrichment levels are interpreted as the relative abundance across two populations having the property of interest. Consider comparing enrichment levels between two prostate cell lines: normal epithelial (PrEC) cells and cancer (LNCaP) cells. There is significant CNV between PrEC and LNCaP cells, as shown in Figure 1A (see also Supplemental Fig. S1). The CNV imbalance leads directly to changes in read density that are not reflective of true changes in methylation (e.g., from MBDCap-seq data). Using Illumina HumanMethylation 450k arrays as an independent assessment of changes in DNA methylation that should be unaffected by CNV (Houseman et al. 2009), Figure 1, B through E, highlights both false-positive and false-negative detections using existing algorithms; these examples are accurately detected by our ABCD-DNA (affinity-based copy-number-aware differential quantitative DNA sequencing analyses) approach (details below). Interestingly, because the prominent copy number state of LNCaP cells is four (Fig. 1A; Supplemental Fig. S1), depth-adjusted read densities are approximately “neutral” (in terms of sampling captured DNA) when LNCaP and PrEC cells have four and two copies, respectively; this further imbalance can be adjusted through “normalization” (adjustments for depth and diversity) in the statistical modeling.

CNV causes false positives and false negatives to various algorithms; ABCD-DNA can recover them. (A) The landscape of CNV between LNCaP (black) and PrEC (gray) cells inferred by PICNIC algorithm (using Affymetrix SNP 6.0 data; see Methods). Using Illumina 450k array data to gauge true differential methylation (see tracks “LNCaP 450k” and “PrEC 450k”), four CNV-induced false-positive (FP) or false-negative (FN) regions in MBDCap-seq data (see tracks “LNCaP_MBD2” and “PrEC_MBD2”) using existing algorithms are shown. Detected differential regions for four methods (ChIPDiff, DiffBind, RSEG, our new approach ABCD-DNA) are shown in black. (B) FN for all algorithms except ABCD-DNA; the change in depth-normalized read density is not particularly strong, but combined with the knowledge that this is a “low” copy region (LNCaP = 2), ABCD-DNA expects fewer reads. Hence, the effective difference is made larger and therefore deemed differential by ABCD-DNA. Similarly, C is amplified in cancer beyond “neutral” (LNCaP = 5), thus ABCD-DNA expects higher read density (if methylated) and correctly increases the effective change. D is similarly amplified, which causes existing algorithms to overstate the differential methylation (i.e., a FP); note the upstream differentially methylated region that all algorithms detect, whereas only ABCD-DNA correctly attributes the downstream change in read density to CNV. (E) Lower copy in LNCaP cells, resulting in lower read depth and FPs for all methods except ABCD-DNA.
There are now a large number of tools for absolute analysis of qDNA-seq data; methods are available for the detection of short distinct events (e.g., MACS) (Zhang et al. 2008), enriched regions (e.g., RSEG) (Song and Smith 2011), ChromaBlocks (Hawkins et al. 2010), or both simultaneously with ZINBA (Rashid et al. 2011)). However, none of the tools are designed explicitly for differential analyses or for when replication is available. Recently, a framework called DiffBind was developed to post-process output from absolute algorithms into merged regions and perform differential analysis based on read densities (Ross-Innes et al. 2012).
A separate class of methods are available to directly detect differential regions, often without the use of input or other control samples (for list of assays and acronyms, see Table 1). For example, Bock et al. (2010) detected changes in read density using Fisher's exact test; CNV is deemed unimportant in their analysis despite no CNV typing. Another strategy, ChIPDiff, assumes beta-binomially distributed tiled bin counts and uses a hidden Markov model (HMM) to combine adjacent differential regions (Xu et al. 2008). Similarly, RSEG scans for differential regions using an HMM with a difference-of-negative-binomials emission distribution (Song and Smith 2011). Other tools are emerging for differential analyses, such as DBChIP (Liang and Keles 2011) or collecting existing Unix-based tools (Bardet et al. 2011), but none of these are explicitly CNV aware. Though specific to DNA methylation, Batman, which transforms read densities into absolute methylation estimates, was recently made CNV aware by first dividing read densities by copy number before differential analysis (Down et al. 2008; Feber et al. 2011). However, this transformation takes measurements off the count scale, which may affect the sensitivity of subsequent statistical analyses.
Table of acronyms for relevant assays and tools
We propose a flexible and general statistical framework called ABCD-DNA that explicitly adjusts for CNV in differential epigenome analyses. First, we describe the statistical framework, which necessarily involves considerations for the estimation of CNV and normalization. Second, we illustrate the effects of CNV on various algorithms for differential analysis across multiple qDNA-seq data sets. By use of independent truth (DNA methylation levels), we demonstrate improved differential detection performance using CNV-aware analyses. Third, we compare the performance of ABCD-DNA and competing methods, demonstrating that the proposed framework is competitive against existing approaches and flexible, irrespective of CNV compensation. All methods are freely available in public software projects, and R scripts to reproduce all analyses are provided.
Results
A general framework for CNV-aware differential qDNA-seq analyses
We propose the following framework:
-
Generate read counts at regions of interest (e.g., at detected peaks, tiled regions genome-wide, or proximal to transcription starts);
-
Estimate copy number offsets from an external data source (see “Copy Number Analyses” below);
-
Estimate normalization offsets based on CNV-neutral loci (See “Normalization” below);
-
Perform differential analysis of count data (e.g., using edgeR) using offsets.
Formally, the strategy for CNV-aware differential analyses can be encapsulated in a generalized linear model (GLM), where
tools applicable to genome-scale data sets have recently become available (Anders and Huber 2010; Zhou et al. 2011; McCarthy et al. 2012). Specifically, let Yij be the read count for region of interest i in sample j (i = 1,…,r and j = 1,…,n where r is the number of regions and n is the number of samples). The read density observed at any
genomic region is modified by systematic effects, such as “effective” sequencing depth, copy number, and underlying biological
factors of interest, as well as sampling and biological variability. Offsets impose a higher or lower expected mean based
on the systematic factors, such as copy number state, depth of sequencing, and sampling rates due to the diversity of the
library sequenced; these are estimated in advance and treated as fixed in the downstream analysis. We model the logarithm
of expected value of Yij as follows: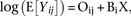 where Oij is an rxn matrix of offsets that match the count matrix, X is an rxk matrix that captures the experimental design (conditions,
covariates), and Bi is a rxk matrix of region-specific coefficients. Oij can be decomposed into log(CNij) + log(1Dj) where CNij is a matrix of offsets for copy number and Dj represents sample-specific offset vector, both of which can be calculated as suggested above. To make inferences regarding
differential enrichment, hypothesis tests can be formulated (e.g., likelihood ratio test) on the parameters of interest within
the Bi matrix (e.g., cancer versus normal); tools for this are readily available (e.g., edgeR) (Robinson et al. 2010a). For specification of all the modeling details (e.g., distributional assumptions, statistical testing), see the Supplemental
Material.
where Oij is an rxn matrix of offsets that match the count matrix, X is an rxk matrix that captures the experimental design (conditions,
covariates), and Bi is a rxk matrix of region-specific coefficients. Oij can be decomposed into log(CNij) + log(1Dj) where CNij is a matrix of offsets for copy number and Dj represents sample-specific offset vector, both of which can be calculated as suggested above. To make inferences regarding
differential enrichment, hypothesis tests can be formulated (e.g., likelihood ratio test) on the parameters of interest within
the Bi matrix (e.g., cancer versus normal); tools for this are readily available (e.g., edgeR) (Robinson et al. 2010a). For specification of all the modeling details (e.g., distributional assumptions, statistical testing), see the Supplemental
Material.
ABCD-DNA can use alternative CNV sources; CNV linearly affects qDNA-seq
ABCD-DNA requires preprocessed CNV information to be delivered to a GLM in a corresponding matrix for regions of interest for each sample; in theory, our approach is independent of the source of CNV information. However, in practice, the success of the CNV adjustment will be determined by the accuracy, resolution, and scale of the CNV estimates, which can vary widely with the platform and preprocessing algorithm used (Curtis et al. 2009). Accuracy should be facilitated by smoothing techniques, such as segmentation (Venkatraman and Olshen 2007), while resolution is ultimately determined by probe spacing (microarrays) or depth of sequencing (HTS). In our analysis of PrEC and LNCaP cells, we used the PICNIC algorithm on Affymetrix SNP 6.0 array data, which resulted in integer-valued CNV estimates due to the homogenous population of the cell lines (Fig. 1A). Supplemental Figure S2 highlights the strong concordance between PICNIC CNV estimates and segmented low-coverage genomic sequencing read densities, after adjusting for GC content and mappability (see Methods). Therefore, only minor differences in the downstream differential analysis between the alternative sources of CNV offsets should result (discussed below). Another important consideration is the scale of the CNV offsets, and specifically, the relationship between CNV and DNA-seq read depths; the GLM model assumes a linear relationship between the offset and expected mean. Supplemental Figure S3 shows M (log-fold-change adjusting for total depth) versus A (average-log-read-density) “smear” plots for three qDNA-seq data sets across PICNIC-defined CNV states, highlighting the increase in M as relative CNV increases. Furthermore, approximate linearity is observed for all qDNA-seq data sets (Fig. 2), which supports the assumption made by ABCD-DNA in conveying such offsets to the GLM model.
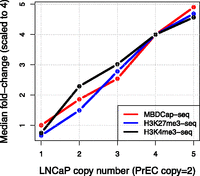
Linearity between CNV and qDNA-seq. Relative read densities scale linearly with CNV for multiple LNCaP/PrEC qDNA-seq (MBDCap, H3K27me3, H3K4me3) data sets. Scaling factors were calculated separately as the median of log-fold changes (median of M-values) for each CNV stratum and each data set (See Supplemental Fig. 3); these medians were exponentiated and scaled according to the most prominent CNV state (L = 4 P = 2). Note that these scaling factors are not actually used in the ABCD-DNA method; they are shown here only to illustrate the relationship between qDNA-seq and CNV.
Normalization to “neutral” regions
qDNA-seq read density at any given locus is affected by biological factors, such as CNV, and technical factors, such as total sequencing depth and library diversity. Therefore, “normalization” is a subtle yet important aspect for allowing accurate comparison of samples. When are read densities comparable, up to a scaling factor? This question has been addressed in the context of RNA-seq data, where not only expression level, but composition of the library and GC content affect read density (Robinson and Oshlack 2010; Hansen et al. 2012). One popular solution is to use a scaling factor (i.e., an offset) called trimmed mean of M-values (TMM), which allows observations to be kept on their original scale (i.e., counts) for statistical modeling. However, TMM normalization does not explicitly handle CNV or the asymmetry of changes in enrichment (e.g., DNA methylation has opposing global loss in cancer and localized gain at CpG-rich regions). To estimate normalization factors, we focus on the most prominent “neutral” state. Typically, this will be genomic regions with two copies. However, as mentioned, most of the LNCaP genome has four copies, so we define neutral as autosomal regions with two copies for PrEC and four copies for LNCaP (Fig. 1A); this spans ∼65% of the reference genome. Figure 3 shows pairwise comparisons of MBDCap-seq samples using only loci from this neutral state. Due to the logarithm transform, variability of M decreases as A increases (Robinson and Oshlack 2010). However, because of differences in composition and global asymmetry in DNA methylation between samples, the center of the M-values does not necessarily occur at zero. Assuming there are regions similarly enriched in both samples, we estimate this bias from “neutral” regions only using the regions of lowest variability (e.g., median of M-values for A > 99th percentile of A-values) (see Fig. 3) and introduce a sample-specific offset into the statistical model to compensate for expected bias in read densities. Support for this strategy is given in Supplemental Figure 4, where normalized data (M-values after adjustment by estimated offsets) for “neutral” loci genome are shown, stratified by CpG density. Despite the asymmetry in DNA methylation, our normalization ensures that the M-value asymptotes are approximately zero, suggesting that read densities are comparable.
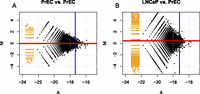
Normalization to “neutral” CNV state using estimated scaling factors. M (depth-normalized log-fold-change) versus A (depth-normalized average-log) “smear” plots for MBDCap-seq data are shown between technical replicates (A) and between cancer and normal (B); each dot represents a 500-bp region of the genome. M is defined as the log-fold-change between two samples (counts divided by library size); A is the average of the log counts divided by library size. (Blue lines) 99th percentile of A-values; (red lines) scale factor estimates (median of M for regions with A greater than 99th percentile).
Differential calls for various assays and algorithms are positively correlated with CNV
Figure 1, B through E, highlights loci where CNV affected read densities, resulting in false or missed detections. To highlight that CNV affects many algorithms genome-wide, we tested several differential approaches: (1) DiffBind coupled with MACS output, (2) RSEG, (3) ChIPDiff, and (4) ABCD-DNA using 500-bp tiled genomic bins. We define relative rate of peak density (RRPD) as the number of regions detected in LNCaP divided by the number detected in PrEC, for each CNV state (Fig. 4). Generally, higher (lower) relative CNV results in more (less) differential region detections, for all algorithms except ABCD-DNA; this positive correlation is indicative of CNV alone affecting the differential calls. Although we do not expect this curve to be completely flat (e.g., interactions between CNV and epigenetics), ABCD-DNA largely removes this association.
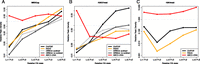
Association between differential peak detection and CNV across LNCaP/PrEC qDNA-seq data sets using various algorithms. The relative rate of peak detection (RRPD), defined as the ratio of the number of regions detected in LNCaP (L) cells to the number of regions detected in PrEC (P), within each CNV stratum is shown for ChIPDiff, RSEG, DiffBind (with and without input subtraction), and ABCD-DNA. DiffBind is based on MACS-detected regions. (A) MBDCap-seq; (B) H3K27me3-seq; and (C) H3K4me3-seq. Due to lack of replication, DiffBind was not run on H3K4me3-seq.
Furthermore, CNV may impact many cancer data sets and algorithms. For example, an independent comparison of the LNCaP and PrEC methylome (Kim et al. 2011) by running a region detection algorithm and simply overlapping lists is strongly affected by CNV (Supplemental Fig. S5). Similarly, differentially methylated regions detected by MeDIP-seq in breast cancer cell lines (Ruike et al. 2010) are associated with CNV, according to their input samples (Supplemental Fig. S6). Taken together, these results suggest that a nontrivial fraction of differential peak detections could be driven simply by CNV, not changes in relative biological enrichment.
CNV offsets improve differential detection performance
To illustrate that the CNV and normalization offsets proposed above can improve differential detection, we use an independent readout of differential methylation on the same LNCaP and PrEC cells. By use of Illumina HumanMethylation 450k BeadChip arrays, DNA methylation estimates at individual CpG sites are summarized as beta values (see Methods). For comparison with the MBDCap-seq data, beta values are averaged over technical replicates and regions of interest. Here, regions of interest comprise nonoverlapping 500-bp tiled genomic segments where 450k probes exist. The averaged beta values are used to label regions as differentially methylated (change in beta > 0.4), not differentially methylated (change in beta < 0.1) or indeterminate (0.1–0.4). GLMs are fitted using the edgeR package with and without CNV offsets (both use normalization offsets), and ranking of regions is according to likelihood ratio test P-values. Other cutoffs for difference in beta values were tested (data not shown), and the results presented here are representative.
Figure 5 shows receiver operating characteristic (ROC) curves for symmetrically chosen truly differentially methylated regions (see Methods), stratified by copy number state, comparing CNV-aware (“ABCD-DNA,” using either SNP arrays or genomic sequencing for CNV offsets) and CNV-unaware GLM strategies (“Naive”); RSEG and DiffBind (with and without input subtraction) are also compared (see Methods). Taken together, these results highlight several features of our new method: (1) Gains in performance can be achieved for non-“neutral” regions; (2) the magnitude of performance gain increases as CNV increases; (3) ABCD-DNA performs equally well, regardless of the source of CNV information (Affymetrix SNP 6.0, low coverage genomic sequencing); and (4) ABCD-DNA outperforms competing methods.
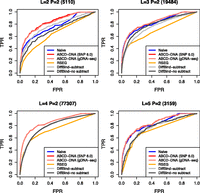
ABCD-DNA outperforms competing approaches. ROC curves (sensitivity versus 1 − specificity) are shown for various differential region detection algorithms operating on MBDCap-seq data, using 450k array data as an independent source of truly and nontruly differentially methylated regions. “Naive” uses offsets to account for (effective) sequencing depth but not CNV; “ABCD-DNA” uses either Affymetrix SNP 6.0 or genomic sequencing to estimate CNV offsets. “RSEG” denotes running rseg-diff with different sensitivity cutoffs. “DiffBind,” which operates on MACS-detected regions, was run both with and without input subtraction. Each panel shows ROC curves for the respective CNV stratum (between LNCaP and PrEC cells), as indicated in the panel title; the number of such regions is shown in parentheses. In the “L = 4 P = 2” panel, Naive and both ABCD-DNA curves almost completely overlap, as do the two DiffBind curves (with and without input subtraction).
To understand the difference that CNV compensation makes genome-wide to differential detection calls, Supplemental Figure S6 gives Venn diagrams showing the overlap of CNV-Aware and Naive calls (adjusted P < 0.01) by CNV state; as expected, differential calls in the “neutral” regions are unaffected, while the overlap degrades significantly as CNV increases. Furthermore, to highlight how ABCD-DNA removes the association between differential detection and CNV, Supplemental Figure S7 shows differential detection Z-scores with and without CNV adjustment, stratified by CNV and by “true” 450k differential status used in the ROC comparisons. Naive scores increase predictably with CNV, whereas ABCD-DNA scores are stable across all CNV states, allowing a better separation of truly differentially methylated from nondifferentially methylated.
Because of the asymmetry in the DNA methylation, ROC comparisons are sensitive to the CNV adjustments made. Probes on the 450k arrays are biased toward CpG-rich regions, and since these regions often gain methylation in cancer, there is a performance advantage to always increasing the log-fold-change, which can confound the interpretation of the CNV compensation. To eliminate this bias, our results above (Fig. 6) used randomly selected truly differentially methylated regions such that the same number increased and decreased. However, Supplemental Figure S8 highlights ROC comparison where this symmetry was not ensured; in this situation, we overstate (understate) performance for lower (higher) relative CNV, as expected.
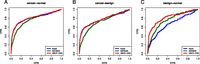
ABCD-DNA outperforms CNV-aware Batman. ROC curves (sensitivity versus 1 − specificity) for three pairwise comparisons are shown for a MeDIP-seq data set (Feber et al. 2011), where Illumina HumanMethylation 27k data is used as an independent source of truly and nontruly differentially methylated regions. “Batman” refers to the CNV-adjusted read densities before running the Batman algorithm and taking differences in methylation estimates. “Naive” refers to a count-based analysis, without accounting for CNV. “ABCD-DNA” refers to a count-based analysis, with additional offsets to account for CNV (estimated from Affymetrix SNP 6.0 data using the PICNIC algorithm). Comparisons are as follows: (A) cancer versus normal; (B) cancer versus benign; and (C) benign versus normal.
ABCD-DNA outperforms CNV-aware Batman
Next, we compared ABCD-DNA against the CNV-aware Batman for the differential analysis of MeDIP-seq data. In the original analysis, read densities were first preprocessed (divided by CNV, explicitly assuming a direct unit slope relationship) to adjust for CNV before using Batman (Feber et al. 2011). Their data set comprises MeDIP-seq, Affymetrix SNP 6.0, and Illumina HumanMethylation 27k arrays for three pooled populations: (1) cancer versus normal (malignant peripheral nerve sheath tumors versus normal Schwann cells), (2) benign versus normal (benign neurofibromas versus Schwann cells), and (3) cancer versus benign. We use the 27k array data as independent “truth” for our performance evaluation (as above, change in beta > 0.4 defines differentially methylated, and change in beta < 0.1 is deemed nondifferentially methylated). We estimated CNV offsets from their Affymetrix SNP 6.0 data using PICNIC and normalization offsets using CNV-neutral regions, as above. Notably, because these are sample mixtures, the CNV estimates could be non–integer-valued. Figure 6 shows ROC curves for the three comparisons using three differential detection approaches: (1) the CNV-aware Batman (“Batman”) (Down et al. 2008; Feber et al. 2011), (2) count-based analysis with only normalization offsets (“Naive”), and, (3) count-based analysis with normalization and CNV offsets (“ABCD-DNA”). Overall, these results suggest that two gains in performance can be made: (1) count-based methods outperform CNV-aware Batman on two out of three comparisons, perhaps suggesting that modeling the data on its count scale followed by direct comparison of read densities performs well; and (2) directly integrating CNV information gives a performance advantage. In addition, Batman is specific to methylated DNA capture assays, whereas ABCD-DNA can be applied to other qDNA-seq assays.
Discussion
CNV affects read densities for various qDNA-seq assays. For differential comparisons between cancer and normal epigenomes, results can be both driven and masked by CNV, thus leading to false positives and reduced power (Fig. 1). Cancer qDNA-seq data sets are on the rise, and many will ultimately be affected by CNV. We present a straightforward solution that explicitly models CNV in a well-established count-based framework. Our method, called ABCD-DNA, estimates CNV and normalization offsets and includes them directly in a GLM, similar to recent approaches applied to RNA sequencing data (Hansen et al. 2012). Thus, we enable a strategy that jointly accounts for effective sequencing depth and CNV, within statistical models that handle biological replication. We verified the approximately linear relationship between CNV and qDNA-seq on multiple cell line data sets, suggesting that offsets are presented on an appropriate scale to modify the mean response.
By use of an independent readout of DNA methylation on two data sets, we demonstrated that ABCD-DNA is competitive against existing differential approaches and integrating CNV through offsets can further improve performance. In addition, the ABCD-DNA framework is flexible and extensible. Because a matrix of offsets is matched to the matrix of read densities, there is a facility for analyzing data sets with sample-specific, possibly noninteger, copy number. For example, patient studies, where each has a different copy number profile, could be analyzed. Furthermore, through the offset matrix, the method can adjust for not only CNV and effective sequencing depth, but other technical factors that affect read density, such as GC content or antibody efficiency (Cheung et al. 2011; Egelhofer et al. 2011; Hansen et al. 2012); further study is required to adequately demonstrate this capability for qDNA-seq data sets. Meanwhile, ABCD-DNA can handle replication and complicated experimental designs, since these are already features of the employed model (McCarthy et al. 2012). In principle, ABCD-DNA can make use of any accurate source of CNV information; however, the success of the CNV adjustment is ultimately reliant on the accuracy, resolution, and scale of these estimates. Furthermore and perhaps most importantly, ABCD-DNA can be applied to differential analysis of various qDNA-seq data sets, including ChIP-seq.
One potential disadvantage of our approach is the reliance on regions of interest, such as regions tiled along the genome; the positioning of these regions could have some effect. An alternative strategy would be to consider overlapping bins tiled at high density, in combination with principled techniques for smoothing, such as HMMs, to assemble differential regions; this work is beyond the scope of the proof of principle presented here. In addition, ABCD-DNA does not currently have a facility for incorporating “input” or control samples; on our evaluation data set, DiffBind's explicit input subtraction did not convincingly improve performance, and other reports have challenged the appropriateness of such controls (Cheung et al. 2011). Further study is required to make general recommendations on this matter.
The main implication of our results is that CNV information, at least for cancer studies, is required for interpretation of qDNA-seq read densities. Failing to account for CNV may result in false positives and false negatives (e.g., Fig. 1B–E) and could have significant impact on downstream analyses. For example, if CNV is responsible for a significant fraction of naively determined differentially enriched regions, downstream analyses, such as functional category analysis or pathway analysis, may be confounded by CNV; that is, enriched pathways may largely be a reflection of CNV, not from changes in the epigenetic factor of interest. Since ABCD-DNA adjusts expected read density by number of copies, the method can also facilitate detection of changes in allele specificity; however, partitioning the reads by allele using genotypes is a more direct approach for this (Statham et al. 2012).
Unfortunately, the requirement for CNV information imposes a potentially costly burden for researchers studying cancer epigenomes, since every sample will need to be CNV typed; this would consume sequencing resources and precious DNA. In practice, the effect of CNV on qDNA-seq can be large or small, depending on the type and severity of the cancers being studied. In the comparison of LNCaP and PrEC cells, the magnitude of CNV change is moderate (most often, changes from four copies to three or five), but a large proportion of the genome (∼35%) is affected, so significant improvements can be made. Depending on the cancer and the severity, copy number aberrations may be larger in magnitude than our data set, and affect larger (or smaller) proportions of the genome (Baudis and Cleary 2001). So, the gains to be made from CNV-aware analyses depend on the data set. However, from our initial results, there is generally only gains to be made after integrating CNV. Furthermore, while the main motivation to develop ABCD-DNA is to compensate for CNV, we have shown that it performs well relative to existing approaches, so the framework may benefit differential qDNA-seq analyses outside of the cancer field.
Methods
Estimating CNV from Affymetrix SNP 6.0 microarrays
The PICNIC tool (Greenman et al. 2010), specifically designed for the analysis of Affymetrix SNP 6.0 arrays, was used to estimate absolute copy number genome-wide using default parameters. These regional estimates were matched to the read densities in tiled bins along the genome and used directly as offsets in the downstream CNV-aware GLM count modeling.
Estimating CNV from genomic sequencing
Since read depths in genomic DNA sequencing are affected by local GC content and mappability, we implemented a R routine in the Repitools package (Statham et al. 2010) called absoluteCN() that calculates read density, GC content, and mappability in bins genome-wide. Bins with mappability <75% are removed; a smooth curve is fit to the mode of depth versus GC content. This relationship is removed for each bin by dividing out the fit at the bin's GC content and then scaled according to knowledge of the most prominent copy state (here, LNCaP = 4 and PrEC = 2). Read densities are then segmented using CBS (Venkatraman and Olshen 2007).
Choosing regions for ROC analysis “symmetrically”
Because the truly differentially methylated regions for the LNCaP versus PrEC comparison are biased toward hypermethylation, we randomly selected the same number of truly hypermethylated and truly hypomethylated regions for the ROC analysis.
ROC analysis using RSEG
To generate ROC curves for RSEG, we ran rseg-diff repeatedly with different values of the -cdf-cutoff parameter (between 0.01 and 0.40). For each of the truly differentially methylated and nondifferentially methylated regions, the score used for ROC analysis was the maximum cdf-cutoff such that the region was deemed differentially enriched, if at all. For descriptions of the commands used for each tool, see the Supplemental website.
ROC analysis using DiffBind
To generate ROC curves for DiffBind, we set a high P-value threshold when calling dba.report(), thus giving scores for the full list of inputted regions. The score used for ranking was the P-value. Furthermore, whether to subtract input reads was controlled by the bSubControl=FALSE argument in the call to dba.analyze(). Otherwise, default parameters were used.
Processing of Illumina HumanMethylation 450k array data
The HumanMethylation 450k arrays were processed using the R/Bioconductor ‘minfi' package using bg.correct = TRUE and normalize = “controls”, to generate beta values. Differences in beta values were used to determine the truly differentially methylated regions.
Reproducibility of analyses and figures in this manuscript
All data and R code used for the generation of figures in this manuscript are available from http://imlspenticton.uzh.ch/robinson_lab/ABCD-DNA/ with further description in the Sweave-based Supplemental Material.
Data sets used
The following data sets (with NCBI Gene Expression Omnibus accession numbers) were used for the main comparisons:
-
MBDCap-seq, Affymetrix SNP 6.0 arrays, and low-coverage genomic DNA sequencing on LNCaP and PrEC cells and MBDCap-seq of SssI (fully methylated DNA; GSE24546) (Robinson et al. 2010c), as well as H3K27me3-seq (GSE38683) and H3K4me3-seq (GSE38682) on the same cell lines.
-
Illumina HumanMethylation 450k bead array on LNCaP and PrEC (GSE34340).
-
From the Feber et al. (2011) study, MeDIP-seq, Affymetrix SNP 6.0 arrays, and Illumina HumanMethylation 27k were available for pools of malignant peripheral nerve sheath tumors, normal Schwann cells, and benign neurofibromas.
Additional analyses to investigate the association between CNV and differential region detection:
-
For Ruike et al. (2010) MeDIP-seq and input-seq data, reads were downloaded from the DDBJ Sequence Read Archive (accession DRP000030) and remapped to the human hg18 genome. A list of differential regions was obtained from Yoshinao Ruike (pers. comm.); analysis of the association between their corresponding input-seq read densities and detected differential regions was performed using a custom R script.
-
For Kim et al. (2011) M-NGS data, the list of differentially methylated regions was obtained from Mohan Dhanasekaran (pers. comm.); using our SNP array data (same cell lines), associations were made to their detected regions using a custom R script.
Data access
A detailed description of the implementation details for ABCD-DNA is given in the Supplemental Material. Software to run ABCD-DNA is freely available within the Bioconductor Repitools package (Statham et al. 2010).
Acknowledgments
We thank Davis McCarthy, Yunshen Chen, and Gordon Smyth for early access to the edgeR GLM code and useful discussions; we thank Andrea Riebler and Alicia Oshlack for reading earlier versions of the manuscript, as well as Rory Stark for helpful discussions and Kate Patterson for help with figures. Funding was from NBCF program (S.J.C.) and NHMRC project grants (C.S., S.J.C.).
Footnotes
-
↵7 Corresponding author
E-mail mark.robinson{at}imls.uzh.ch
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139055.112.
Freely available online through the Genome Research Open Access option.
- Received February 13, 2012.
- Accepted August 17, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








