
Contributions of mRNA abundance, ribosome loading, and post- or peri-translational effects to temporal repression of C. elegans heterochronic miRNA targets
- 1Department of Genetics,
- 2Department of Pathology, Stanford University, Stanford, California 94305-5324, USA
Abstract
miRNAs are post-transcriptional regulators of gene activity that reduce protein accumulation from target mRNAs. Elucidating precise molecular effects that animal miRNAs have on target transcripts has proven complex, with varied evidence indicating that miRNA regulation may produce different molecular outcomes in different species, systems, and/or physiological conditions. Here we use high-throughput ribosome profiling to analyze detailed translational parameters for five well-studied targets of miRNAs that regulate C. elegans developmental timing. For two targets of the miRNA lin-4 (lin-14 and lin-28), functional down-regulation was associated with decreases in both overall mRNA abundance and ribosome loading; however, these changes were of substantially smaller magnitude than corresponding changes observed in protein abundance. For three functional targets of the let-7 miRNA family for which down-regulation is critical in temporal progression of the animal (daf-12, hbl-1, and lin-41), we observed only modest changes in mRNA abundance and ribosome loading. lin-41 provides a striking example in that populations of ribosome-protected fragments from this gene remained essentially unchanged during the L3–L4 time interval when lin-41 activity is substantially down-regulated by let-7. Spectra of ribosomal positions were also examined for the five lin-4 and let-7 target mRNAs as a function of developmental time, with no indication of miRNA-induced ribosomal drop-off or significant pauses in translation. These data are consistent with models in which physiological regulation by this set of C. elegans miRNAs derives from combinatorial effects including suppressed recruitment/activation of translational machinery, compromised stability of target messages, and post- or peri-translational effects on lifetimes of polypeptide products.
The Caenorhabditis elegans heterochronic genes lin-4 and let-7 (Lee et al. 1993; Wightman et al. 1993; Reinhart et al. 2000) were the first reported members of the family of regulatory noncoding RNAs known as microRNAs (miRNAs). These 21–22-nucleotide (nt) RNAs were found to act through partially complementary nucleotide sequences in the 3′ untranslated regions (UTRs) of target mRNAs to prevent the accumulation of protein product at specified developmental time points. Subsequent studies revealed that miRNAs are present in the genomes of species residing in all eukaryotic kingdoms (Pasquinelli et al. 2000; Griffiths-Jones et al. 2008) and constitute an ancient and broadly conserved gene regulatory mechanism.
Animal miRNAs have been the focus of intense study in the past decade (Bartel 2009), leading to recognition of several generalized features of these molecules. Salient features shared by a number of well-characterized miRNAs, although not necessarily expected to be universal, include: (1) Canonical miRNAs are genomically encoded as long precursor transcripts, which are processed by two endonucleolytic steps into ∼22-nt mature miRNAs (Lee et al. 2003, 2004); (2) miRNAs generally bind target mRNAs at positions that are only partially complementary to the nucleotide sequence of the miRNA, with targeting efficacy variably influenced by contextual features that are still not fully understood (Ha et al. 1996; Vella et al. 2004a,b; Didiano and Hobert 2006; Shin et al. 2010; Garcia et al. 2011); (3) miRNAs are associated both physically and functionally with effector proteins of the Argonaute family (Huntzinger and Izaurralde 2011).
While considerable progress has been made characterizing miRNA repertoires and elucidating their physiological roles, the molecular mechanism(s) by which miRNAs (and their effector proteins) reduce protein output of target genes have remained controversial. Initial analysis of the C. elegans miRNAs lin-4 and let-7 indicated that miRNAs could direct substantial regulation of target protein levels under conditions where steady-state levels of target mRNAs did not appreciably decrease (Olsen and Ambros 1999; Seggerson et al. 2002). Additionally, polysome sedimentation profiles of target mRNAs were shown to be similar in the presence and absence of the targeting miRNA, leading to the suggestion that the C. elegans heterochronic miRNAs repress translation at a step after translation initiation. In a more recent study of lin-4 and let-7 repression in C. elegans, a somewhat larger decrease in target mRNA levels has been reported (Bagga et al. 2005). Subsequent analysis of mRNA abundance changes by the same group suggested that the observed loss of target mRNAs depends on specific growth conditions (Holtz and Pasquinelli 2009). Detailed studies of genetic and biochemical requirements in specific silencing models for C. elegans have suggested a number of additional (and non-mutually exclusive) possibilities for level-of-action by regulatory miRNAs, including translation initiation (Ding and Grosshans 2009) and mRNA deadenylation (Wu et al. 2006).
Experiments in other animal systems have similarly provided support for a variety of possible mechanisms for miRNA action. At the translation level, reports have indicated that miRNAs can inhibit target translation by blocking translation initiation (e.g., Pillai et al. 2005; Mathonnet et al. 2007; Thermann and Hentze 2007; Djuranovic et al. 2012), blocking 60S subunit joining (Wang et al. 2008), inducing premature ribosome drop-off (e.g., Petersen et al. 2006), or co-translational or peri-translational proteolysis (Nottrott et al. 2006). Considerable evidence has also accumulated to indicate that target mRNA deadenylation and subsequent degradation can be an important, or even the primary, means of miRNA-mediated silencing (Rehwinkel et al. 2005; Behm-Ansmant et al. 2006; Giraldez et al. 2006; Wu et al. 2006; Hendrickson et al. 2009). Proteomic analyses have reached diverse conclusions about the degree to which miRNAs affect mRNA levels and protein production (Baek et al. 2008; Selbach et al. 2008; Jovanovic et al. 2012).
High-throughput ribosome profiling is a powerful and sensitive method for assessing the translational activity of mRNAs. This technique was introduced by Ingolia et al. (2009) and extends a classical technique developed by Steitz (1969) to a genome-wide scale, potentially generating position-specific maps of ribosome occupancy for all expressed mRNAs. The various models proposed for miRNA-mediated translational repression offer specific predictions for the ribosome occupancy patterns of target mRNAs. If translation initiation is blocked or reduced, we would expect to observe a lower overall abundance of ribosome-protected fragment (RPF) reads for a transcript, but would not necessarily expect a change in the distribution of ribosomes along the length of the transcript. Inhibited elongation might be expected to produce “pause sites” represented by high RPF densities at specific locations, or could result in a buildup of ribosome density at the 5′ ends of mRNAs if elongation is substantially but nonspecifically inhibited, producing a ribosomal “traffic jam.” Premature ribosome drop-off would result in a similar pattern, with a shift in read density toward the 5′ end resulting from decreasing ribosome occupancy toward the 3′ ends of target mRNAs. Other, more complex shifts affecting both initiation and translation are also conceivable, and we note that such concurrent changes could produce varied patterns of RPFs.
Several groups have applied ribosome profiling to the study of miRNA action. Guo et al. (2010) applied the technique to cultured mammalian cells, analyzing the results of both transfection with exogenous miRNAs and knockout of an endogenous miRNA. They report that for most targets the decrease in protein levels could be explained by changes in mRNA levels, observing no significant changes to the ribosome profiles of regulated mRNAs. In contrast, application of ribosome profiling by Bazzini et al. (2012) to examine broad miR-430-dependent repression of expression in zebrafish embryos suggested that miR-430 regulation in this context results in inhibition of translation that precedes mRNA decreases. Although these are valuable experimental systems, neither system offers the possibility of examining individual miRNA–target interactions with a known physiological consequence. Given the likely diversity of downstream effects, it is of considerable interest to examine the behavior of miRNA targets in cases where individual interactions have a defined biological role.
The C. elegans heterochronic pathway is an amenable model for studying the functional consequences of regulation by animal miRNAs, with rigorous genetic and molecular analysis providing for definitive assignment of several target genes as functionally down-regulated by direct miRNA interaction during development (Fig. 1A): The lin-4 miRNA is responsible for down-regulating the lin-14 and lin-28 mRNAs at the transition between larval stages L1 and L2, with lin-4 loss-of-function mutations resulting in reiteration of L1 cell fates (Lee et al. 1993; Wightman et al. 1993; Moss et al. 1997); the trio of let-7 family members miR-48, miR-84, and miR-241 down-regulate the daf-12 and hbl-1 mRNAs at the L2/L3 transition (Abrahante et al. 2003; Lin et al. 2003; Abbott et al. 2005; Grosshans et al. 2005; Hammell et al. 2009), and let-7 targets lin-41 at the L3/L4 transition (Reinhart et al. 2000). Some degree of overlap exists in the pathway in that let-7 likely targets daf-12 and hbl-1 in L4 stage animals (Abrahante et al. 2003; Grosshans et al. 2005). Additionally, regulation of the heterochronic genes is subject to multiple inputs (e.g., Morita and Han 2006), including transcriptional control, but functional down-regulation is dependent on controlling miRNAs. Collectively, the five mRNAs known to be targeted at specific developmental time points by miRNAs provide a valuable case study for miRNA function in a physiological context.
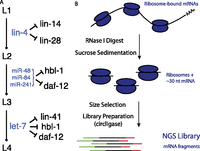
miRNAs in the C. elegans heterochronic pathway. (A) Five genes of the heterochronic pathway are known to be targeted by miRNAs during C. elegans larval development. (B) Overview of the high-throughput ribosome profiling procedure. (NGS) Next generation sequencing.
Focusing on the heterochronic miRNA targets, we have applied immunoblotting, mRNA-seq, high-throughput ribosome profiling, and an allele-specific mRNA counting approach to C. elegans larvae to determine the molecular consequences of regulation by a set of miRNAs on natural targets in a developing animal.
Results
We performed parallel mRNA-seq and ribosome profiling on developmentally synchronized populations of wild-type C. elegans early L1, L2, L3, and L4 larval stage animals, following a procedure adapted from the one described by Ingolia et al. (2009) (Fig. 1B). We stress that the approach here has been applied to whole-animal samples, with the advantage of allowing analysis of interactions in a true in vivo setting, but with the disadvantage of preventing analysis of potential tissue-specific effects (C. elegans cannot be dissected on a scale that would allow cell-type-specific analysis). Importantly, molecular phenotypes (decreased protein and/or mRNA abundances) resulting from lin-4 and let-7 action have previously been observed with whole-animal approaches (e.g., Olsen and Ambros 1999; Bagga et al. 2005), indicating that regulatory phenomena associated with the heterochronic miRNAs are resolvable even when using a mixture of tissues.
We analyzed ribosome profiling and mRNA-seq libraries from a minimum of two biological replicates at each stage, with an additional technical replicate for each time point (six biological replicates were analyzed for L1, three for L4). A subset of these libraries have been described previously in the context of associating codon choice and translation elongation rates; remaining libraries were constructed from staged C. elegans populations and validated as described by Stadler and Fire (2011). For each library of RPFs, we confirm alignment to the C. elegans transcriptome and the overwhelming enrichment for coding regions over 5′ and 3′ UTRs (Ingolia et al. 2009). mRNA-seq libraries, prepared using an identical scheme but using fragmented naked poly(A)+ RNA (rather than ribosome-protected nuclease digest), were enriched for sequences in mature mRNA. As expected, the mRNA-seq libraries show no exclusion of 5′ and 3′ UTR segments.
Assessment of protein and mRNA levels
In our initial analysis, we queried mRNA-seq and immunoblotting data to obtain relative steady-state levels of protein and mRNA as a function of larval stage.
lin-4 miRNA, rare or absent on hatching, accumulates at high levels in L2 animals (Lee et al. 1993). Prior immunoblotting experiments estimated a decrease in protein levels between five- and 20-fold for the lin-4 targets in L2 animals (12–15-fold for LIN-14 [Olsen and Ambros 1999]; 10–20-fold for LIN-14 and LIN-28 [Seggerson et al. 2002]; fivefold for LIN-14 [Holtz and Pasquinelli 2009]). To augment studies in the literature, we carried out immunoblotting experiments for LIN-14 and LIN-28 using staged populations prepared as above as starting material (Supplemental Fig. S1). Multiple samples (including samples used for the mRNA and ribosome sequencing analysis), technical replicates, and normalizations were used for this analysis, with linearity of assays confirmed through a series of sample mixing experiments (see Supplemental Material). These experiments confirmed earlier results from several of the initial lin-14 and lin-28 studies (Olsen and Ambros 1999; Seggerson et al. 2002), showing an 18-fold decrease for LIN-14 between L1 and L2 (36-fold L1 to L4; Supplemental Fig. S1A) and a 14-fold decrease in LIN-28 abundance (>100-fold L1 to L4; Supplemental Fig. S1B).
Various normalization methods can be used for analysis of RNA sequencing data (Mortazavi et al. 2008); we use the trimmed mean of the M-values (TMM) method from the edgeR software package (Robinson and Oshlack 2010). In brief, the TMM method normalizes the data in order to minimize the variance of the fold-changes between multiple samples. TMM normalization is used in presenting the results below, with supplementary analysis demonstrating similar conclusions with alternative normalizations presented in Supplemental Figure S2.
For the lin-4 targets lin-14 and lin-28, our mRNA-seq data indicate modest decreases in steady-state mRNA levels coinciding with accumulation of lin-4 (Fig. 2A, L1–L2; Supplemental Table S1). lin-14 mRNA decreased approximately threefold between L1 and L2 (P = 0.001), then showed a reproducible increase in L3 animals before returning to L2 levels in L4. lin-28 mRNA levels similarly decreased approximately threefold (P = 0.001) between L1 and L2 and remained relatively constant throughout subsequent larval development. These modest changes are consistent with a subset of earlier analyses with Northern blot hybridization in which the targeted mRNAs were still readily detected by hybridization but appeared to be less concentrated (Wightman et al. 1993).
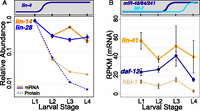
Changes in mRNA levels of miRNA targets measured by mRNA-seq. (A) Steady-state mRNA levels measured by mRNA-seq (solid lines) and protein levels (dashed lines) measured by immunoblotting for lin-4 targets (lin-14 and lin-28). mRNA and protein levels were normalized to L1 levels, and relative levels at subsequent larval stages were plotted on the same scale with a logarithmic vertical axis. (B) mRNA levels measured by mRNA-seq for let-7 and miR-48/miR-84/miR-241 targets (lin-41, daf-12, and hbl-1) for each larval stage, with nonlogarithmic vertical axis. Counts were normalized using the TMM method in the EdgeR package (Robinson and Oshlack 2010) to account for both library size and composition differences. (Data points) Average normalized tag count for all replicates at each stage; (error bars) standard error of the mean. Significance of differential mRNA levels across larval development (L1–L4) was determined by a two-sided t-test: lin-14 P = 0.07, lin-28 P = 0.05, lin-41 P = 0.21, hbl-1 P = 0.001, daf-12 P = 0.02. Additional normalizations are described in Supplemental Figure S2.
A distinct group of miRNAs helps to orchestrate the L2/L3 transition. At that point, proper developmental progression includes the down-regulation of two genetically defined targets, hbl-1 and daf-12, by three members of the let-7 family (mir-48, miR-84, and miR-241) (Abbott et al. 2005; Hammell et al. 2009). Steady-state mRNA measurements for hbl-1 and daf-12 exhibited little if any change between L2 and L3 stages, with an apparently more significant decrease in L4 (Fig. 2B; Supplemental Table S1). The observed decrease during the L3/L4 transition in these experiments was ∼2.7-fold for daf-12 (P = 0.07) and approximately fivefold for hbl-1 (P = 0.07). It is notable that decreased mRNA levels for these let-7 family targets are not evident until L4, while protein levels are reduced in L3 (Abbott et al. 2005; Hammell et al. 2009).
From previously published analyses, lin-41 appears to be primarily a target of let-7 (Abbott et al. 2005). lin-41 mRNA levels did not change significantly in the larval stages examined (Fig. 2B; Supplemental Table S1).
We next used lin-14 as a test case to ask whether decreases in mRNA levels were direct consequences of miRNA-interacting sequences in the 3′ UTR. The lin-14(n536) allele contains a large 3′ UTR deletion that eliminates five of the seven lin-4 binding sites (Fig. 3A; Ambros and Horvitz 1987; Wightman et al. 1993). The lin-14(n536) allele by itself is insensitive to lin-4 regulation and behaves as a gain-of-function mutation, reiterating early cell fates at later stages, the same phenotype observed in lin-4 loss-of-function alleles. Although this deletion provides a potentially lin-4-resistant target for comparison, the substantial developmental defects in n536-carrying animals complicate any equivalency assumptions in comparing expression to nonmutant lin-14 in wild-type animals. Toward an unbiased comparison, we generated a heteroallelic configuration in which a single functional copy of lin-14 is provided to animals, development is superficially normal, and expression can be compared in the same cell between normal and 3′-deleted alleles. The heteroallelic construction relies on a revertant chromosome in which a missense mutation in cis to the 3′ deletion produces a miRNA-resistant transcript that behaves as a recessive loss-of-function mutation. (Ambros and Horvitz 1987; Reinhart and Ruvkun 2001). The revertant chromosome, n536n539, carries the n536 3′ deletion, as well as a second point mutation in the coding region. lin-14(n536n539)/+ hermaphrodites are thus phenotypically wild type (develop normally) and contain one lin-4-sensitive allele and one lin-4-insensitive allele of lin-14. These alleles can be distinguished by sequence analysis of the region containing the point mutation.
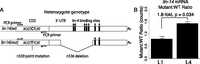
lin-14 mRNA abundance in animals heterozygous for a 3′ UTR deletion. (A) Schematic of wild-type and mutant lin-14 alleles in the heteroallelic configuration. The n536 deletion is confined to the 3′ UTR and eliminates five lin-4 binding sites. The n539 point mutation is used to distinguish mutant from wild-type transcript; reverse transcription and PCR primers were selected using regions around the point mutation that are identical in both alleles, generating products of identical length and differing only at a single internal position. (B) Abundance ratios for wild-type and mutant alleles in L1 and L4 larvae, as measured by Illumina sequencing. Individual ratios were measured as the simple ratio of mutant to wild-type reads, error bars represent standard error of the mean, P-value is from a two-sided t-test. Equivalent data derived using Sanger sequencing is shown in Supplemental Figure S3.
We used both bulk (Sanger) sequence analysis and high-throughput Illumina sequencing to characterize mRNA ratios in lin-14(n536n539)/+ animals. Total nucleic acid was purified from populations (five animals) of early L1 and mid L4 stages. Selecting gene-specific primers not affected by either mutation, we performed reverse transcription and subsequent PCR amplification of the region surrounding the n539 point mutation. Reconstruction experiments with mixed populations of DNA were used to test for and confirm the equivalent amplification of the two alleles (Supplemental Fig. S3).
Analysis of bulk products by Sanger sequencing indicated approximately equivalent levels of the two allelic mRNAs in L1 heterozygotes, but an increase in the ratio of mutant to wild-type transcript in L4 animals, equivalent to a 1.8-fold decrease in wild-type mRNA levels (Supplemental Fig. S3; Supplemental Table S2A). High-throughput sequencing analysis of the same populations of molecules yielded highly similar results (1.8-fold, P = 0.03), confirming a developmental increase in the ratio of mutant to wild-type lin-14 mRNA (Fig. 3B; Supplemental Table S2B). This result implicates the sequence in the lin-14 3′ UTR covered by the n536 deletion as responsible for a portion of the decrease in lin-14 mRNA levels between early- and late-stage animals. Our mRNA-seq measurements recorded a 4.5-fold decrease in lin-14 between L1 and L4. The differences between these measurements may be accounted for by the two remaining lin-4 binding sites in the n536n539 3′ UTR, transcriptional regulation, or may reflect differences in stability of the two transcripts.
Measurement of ribosome-protected fragment abundance and ribosome loading
Because each RPF corresponds to a single ribosome, the fraction of the pool of RPFs derived from a given mRNA is an effective surrogate for the fraction of cellular ribosomes bound to that message. Counting RPF reads thus provides a way to assess the number of ribosomes bound to an mRNA and consequently provides a metric for the translational activity of a gene.
With the onset of lin-4 regulation in L2, both targets of lin-4 show decreases in RPF abundance that closely mirror the decreases observed for mRNA abundance (∼2.6-fold for lin-14, ∼3.9-fold for lin-28) (Fig. 4A; Supplemental Table S3). This decrease becomes more pronounced in later larval development, with total decreases of ∼7.4-fold for lin-14 and ∼10-fold for lin-28 from L1 to L4.
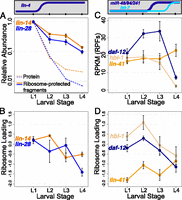
Changes in ribosome-protected fragment abundance and ribosome loading for heterochronic miRNA targets. Total RPF levels and ribosome loading at each larval stage are shown for lin-4 targets lin-14 and lin-28 (A–B) and let-7 family targets hbl-1, daf-12, and lin-41 (C–D). Count normalization and axis scaling are as in Figure 2, and protein levels are plotted for LIN-14 and LIN-28 as a reference (protein data are the same as in Fig. 2). Ribosome loading is defined as the log2 ratio of normalized counts from RPF libraries to corresponding mRNA-seq libraries. The significance of changes in RPF abundance across larval development (L1–L4) was determined by a two-sided t-test: lin-14 P = 0.003, lin-28 P = 0.01, lin-41 P = 0.29, hbl-1 P = 0.01, daf-12 P = 0.002. (Error bars) Standard error of the mean.
The ratio of the proportion of an mRNA in the RPF pool (e.g., Fig. 4A) to its proportion in the poly(A)-selected mRNA pool (e.g., Fig. 2A) serves as one measure of mRNA–ribosome association. Explicitly, we refer to the log2 ratio of RPF counts to mRNA-seq counts as “ribosome loading.” As lin-4 accumulates to high levels in L2, ribosome loading of both target mRNAs remains largely constant (decreases in RPFs parallel decreases in mRNA abundance), whereas in later stages ribosome loading decreases (RPF levels decrease while mRNA abundance remains mostly unchanged) (Fig. 4B; Supplemental Table S4).
Though daf-12 and hbl-1 are targeted by the mir-48/84/241 cluster beginning in late L2, we observe that RPF levels for these mRNAs do not appreciably decrease at the L2–L3 transition, but decrease significantly in L4 larvae (L2–L4 decreases of ∼8.3-fold, P = 0.017 for hbl-1, ∼4.5-fold, P = 0.001 for daf-12) (Fig. 4C; Supplemental Table S3). mRNA levels for daf-12 and hbl-1 actually increase slightly in L3 larvae (Fig. 2B); for these genes, ribosome loading declines modestly in L3, followed by a more substantial decrease in L4 for both targets (Fig. 4D; Supplemental Table S4).
lin-41 exhibited an unexpected pattern of RPF abundance (Fig. 4C; Supplemental Table S3), with a very small and not significant increase in RPF levels in L4 larvae. This was unexpected, as the L3/L4 transition corresponds to the onset of strong let-7 expression and dramatic down-regulation of LIN-41 protein levels in reporter assays (Reinhart et al. 2000). Ribosome loading for lin-41 increased ∼1.6-fold between the L3 and L4 stages (Fig. 4D; Supplemental Table S4).
We considered the possibility that, due to potential experimental or environmental variables, lin-41 down-regulation had not yet occurred in our populations of L4 animals. As a read-out of LIN-41 activity, we investigated the status of the lin-29 mRNA. LIN-29 is a zinc-finger transcription factor whose accumulation in late L4 is necessary for progression to the adult stage (Rougvie and Ambros 1995; Bettinger et al. 1996; Slack et al. 2000). LIN-41 is required for translational repression of LIN-29 expression, and the down-regulation of LIN-41 levels by let-7 is necessary to allow expression of LIN-29 (Slack et al. 2000). Examining lin-29 in our ribosome profiling and mRNA-seq data shows that, while lin-29 mRNA levels remain relatively constant between L3 and L4, lin-29 ribosome loading is increased ∼10-fold (Supplemental Fig. S4). This result strongly suggests that LIN-41 activity has been down-regulated in our L4 samples, and that the consequences of this regulation are resolvable using a whole-animal approach.
Examining high-resolution profiles of ribosome elongation
The earliest mechanistic studies of C. elegans miRNAs (Olsen and Ambros 1999; Seggerson et al. 2002) demonstrated that lin-4-repressed mRNAs could maintain a ribosome-dependent sedimentation in the heavy fractions on a sucrose gradient, suggesting the possibility of post-initiation inhibition of translation. Ribosome profiling provides data on ribosome occupancy by position within each transcript, allowing us to test several specific hypotheses regarding post-initiation means of translational repression. Visual examination of ribosome occupancy profiles of annotated targets of lin-4 and let-7 family miRNAs at stages before and after the onset of miRNA regulation failed to reveal dramatic differences (Fig. 5). In particular, we observed no evidence for strong, reproducible ribosome pause sites within the coding region, which might result from site-specific inhibition of translation elongation. We similarly did not observe a shift in ribosome density toward the 5′ and away from the 3′ end, which we would expect to result from premature ribosome drop-off or traffic jams produced by nonspecific inhibition of elongating ribosomes.
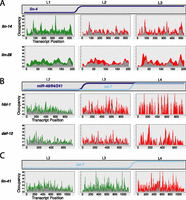
Ribosome occupancy profiles of miRNA targets. Stage-specific ribosome occupancy profiles are shown for targets of lin-4 (A), miR-48/miR-84/miR-241 (B), and let-7 (C). Occupancy profiles are generated by first assigning counts to each codon position in a transcript based on the number of RPF reads whose P-site falls on that codon (RPFs contain 12 nt 5′ to the P-site, see Ingolia et al. 2009; Stadler and Fire 2011). Occupancy values are then normalized within each data set by dividing the counts value at each position by the total counts for that transcript, then multiplying by the transcript length, such that the average codon value is one. Finally, individual profiles are smoothed using kernel density estimation. Data integrated from all replicate data sets are shown: The top of the gray region represents the minimum value observed at that position among all replicates, while the top of the colored region represents the maximum value observed. Equivalent non-smoothed and non-normalized plots are shown in Supplemental Figure S7.
Despite the absence of any dramatic differences, we considered the possibilities of subtle differences in the ribosome occupancy profile that may indicate fundamental differences in interaction with the translational machinery. In this context, it was of interest to determine whether observed variations in profile were beyond the levels expected for biological or experimental noise; specifically whether the occupancy profiles for heterochronic miRNA targets exhibited greater variability than observed for the vast majority of genes with no known heterochronic connection. To provide such a comparison, we divided each transcript into eight bins of equal length, determining and comparing the relative density of RPF reads within each bin at regulated and unregulated stages. Each defined miRNA target could thus be compared to other transcripts of similar coverage levels for the degree of temporal variation/noise. As is evident from these comparisons (Fig. 6), gross changes in ribosome occupancy profiles overwhelmingly fell within the range observed for the background population of transcripts.
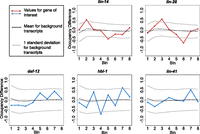
Ribosome occupancy profiles: gross changes. Each gene was divided into eight bins of equal length, and the relative occupancy of RPFs within each bin was calculated as a proportion of the total reads mapping to the gene within the sample that map to the bin. Occupancy for each bin was then compared between miRNA-unregulated and -regulated stages for miRNA targets (L1–L2 for lin-14 and lin-28, L2–L4 for hbl-1, daf-12, and L3–L4 for lin-41). The change for a given bin is the simple difference (regulated − unregulated), such that a positive score indicates the bin had greater occupancy in the miRNA-regulated stage. (Gray bars) Standard deviation of bin changes for mRNAs of similar coverage, with similar coverage defined as those genes with reads-per-codon values within 20% of the mRNA of interest.
In a second, related analysis, we used a sliding-window approach to derive a metric for positional comparison between conditions. In brief, the normalized RPF read occupancy was determined within a set of equally sized sliding windows on each mRNA in the transcriptome and compared between stages corresponding to miRNA-regulated and unregulated time points for our genes of interest. These comparisons yield a distribution of difference scores for each mRNA (positional difference score, see Methods). Comparison of the features of these distributions (mean, median, and maximum difference scores) showed that the changes in ribosome occupancy profiles for miRNA targets are in the range expected for nonheterochronic mRNAs of similar coverage (Supplemental Fig. S5).
A third comparison of profiles employed coincidence statistics. These analyses detect substantial differences between mRNA-seq and RPF profiles. No significant differences in profile were detected in comparing coincidence values between samples at different developmental stages (see Methods; Supplemental Table S5).
Taken together, these results indicate that ribosome occupancy profiles do not change dramatically on miRNA-repressed mRNAs.
Adenylation status of miRNA targets
Numerous studies have provided evidence for deadenylation of target mRNAs as a consequence and/or cause of miRNA-mediated silencing (Rehwinkel et al. 2005; Behm-Ansmant et al. 2006; Giraldez et al. 2006; Wu et al. 2006). Persistence of deadenylated transcripts will likely vary between biological systems and between cell types and conditions within a system, thus some systems may preserve a population of deadenylated mRNA and some may not. To test for the presence of stable deadenylated miRNA target transcripts in C. elegans, we performed RNA-seq on fragmented total RNA [without performing poly(A) selection] from L1 and L4 stage wild-type animals, time points at which targets of lin-4 and let-7 family miRNAs are active and repressed, respectively. We compared tag counts from these total RNA sequencing experiments with counts from poly(A)-selected mRNA-seq from corresponding larval stages with the expectation that mRNAs that are completely (or near-completely) deadenylated will be depleted from the poly(A)-selected pool and enriched in relative abundance in total RNA sequencing libraries.
As an assessment of the efficacy of this methodology, we examined replication-dependent histone genes, mature forms of which generally lack poly(A) tails (Marzluff 2005; Mangone et al. 2010; Jan et al. 2011). These mRNAs were significantly more enriched (∼10–20-fold) in total RNA compared to poly(A)+ selected RNA (Supplemental Fig. S6A,B). Several additional nonhistone mRNAs appeared grouped with these histone genes, indicating that these genes may be strongly deadenylated or may utilize alternative 3′ end structures. While this set of genes demonstrates that this methodology can resolve poly-adenylated vs. non-poly-adenylated transcripts, we have not explored a potential nonlinear relationship between mean poly(A) length and capture by oligo-dT, leaving open the possibility that changes in poly(A) tail length (short of near-complete deadenylation) may not be detected by this comparison.
Using the same criteria that detected the replication-dependent histones, we observed no evidence for extensive deadenylation of lin-4 or let-7 family regulated targets, as these transcripts do not show a significant trend toward higher abundance in the total RNA pool (Supplemental Fig. S6C).
Discussion
Early investigations of the fates of C. elegans miRNA targets established the initial paradigm for the mechanism of animal miRNA action; specifically, that miRNAs direct translational repression. Subsequent work in a variety of systems has pointed to a more diverse array of potential regulatory outcomes. From the evolving literature, it has remained possible (and perhaps likely) that animal miRNAs can direct various molecular outcomes depending on the identity of the specific miRNA, cell or tissue type, developmental stage, cell cycle stage, or species. In this work, we attempt to define the molecular effects of regulation by a limited set of well-characterized miRNAs in the specific physiological context of C. elegans larval development.
Our work involves both conventional and high-throughput “next generation” assays, including ribosome profiling. We note two important characteristics of ribosome profiling data: (1) RPF abundances (and by extension, ribosome loading measurements) are measured relative to total RPFs in a given sample and are thus relative and not absolute; global changes in translational efficiency affecting all mRNAs would not be detected. (2) Regulatory mechanisms in which translationally inhibited mRNAs are rapidly degraded could result in changes evident much more prominently in mRNA level than ribosome loading (Hu and Coller 2012).
Four conclusions come from our observations
-
(1) Levels of target mRNA decrease as the animals progress through larval development, with a fraction of this difference (assayed for lin-14) dependent on miRNA-interacting regions of the 3′ UTR.
-
(2) We observe, for some targets, an overall decrease in the degree of ribosome loading as the animals progress to late larval stages, potentially accounting for a fraction of functional down-regulation.
-
(3) Ribosomal profiles from target mRNAs during down-regulation showed no evidence for either premature ribosomal drop-off or long-term ribosomal pausing as the underlying mechanism of miRNA-triggered inhibition.
-
(4) The modest differences in observed ribosome loading and mRNA levels for the two miRNA targets for which we had definitive protein data (the lin-4 targets LIN-14 and LIN-28) were insufficient to account for much more dramatic differences in target protein abundance.
For the two targets of lin-4 and for both targets of the miR-48/miR-84/miR-241 group, we observe decreases in the amount of ribosome-associated mRNA between early (L1) and late (L4) larval stages. In these cases, comparison of mRNA abundance measurements and RPF levels shows that the decrease results from a combination of decreased ribosome loading and decreased mRNA abundance.
Comparing our ribosome profiling data to measurements of protein levels and known staging of biological function in the action of the relevant inhibitory miRNAs reveals evidence for additional regulation. Specifically, lin-14 and lin-28 total RPF levels decrease only approximately three- to fourfold between the early L1 and L2 stages, while protein levels between these samples decrease 18- and 14-fold for LIN-14 and LIN-28, respectively. A difference between RPF level and protein accumulation is seen over the course of larval development, as protein decreases between L1 and L4 substantially exceed the decrease in RPFs for both LIN-14 and LIN-28 (cf. Supplemental Table S3 and Supplemental Fig. S1).
Likewise, decreases in RPF abundance for hbl-1 and daf-12 show an apparent temporal delay, as levels remain constant across the L2/L3 transition, though the miR-48/miR-84/miR-241 miRNAs accumulate and repress these mRNAs beginning in late L2 (Abbott et al. 2005; Hammell et al. 2009), and only decrease beginning in L4 animals. Most strikingly, lin-41 RPF levels are unchanged in L4 larvae, despite strong repression by let-7 in this stage.
The lack of concordance between changes in RPF levels and protein accumulation leaves open the possibility of a primary regulatory event that may precede or operate independently of the destabilization of the mRNA or changes in ribosome association. Our observation that ribosome occupancy profiles do not appreciably change with the onset of miRNA regulation renders several models of post-initiation translational repression unlikely, such as premature ribosome drop-off or site-specific pausing of elongating ribosomes. An intriguing possibility would be the simultaneous transcript-wide “freezing” of bound ribosomes, potentially through transport to a cellular compartment in which translational capacity is low. Alternative models in which miRNA-inhibited transcripts are translated but produce polypeptide products subject to early degradation are also consistent explanations.
Our data thus define a set of miRNA-dependent regulatory events which cannot be accounted for by single-factor models of mRNA destabilization, compromised translation initiation, or inhibition of translation elongation. Instead, these miRNAs appear to contribute to C. elegans developmental progression through mechanisms that may include, but which also extend beyond, these three alternatives.
Methods
Samples
C. elegans N2 animals were cultured as described in Brenner (1974). Animals were allowed to hatch in the absence of food for 24 h at 16°C, producing synchronized populations of L1 stage larvae. L1s were transferred to enriched agar plates with Escherichia coli OP50 food source at 16°C for 4 h (early L1), 34 h (L2), 45 h (L3), or 63 h (L4). Animals were harvested by washing and flash-freezing aliquots in liquid nitrogen. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR).
Ribosome profiling and library preparation
Profiling and library preparation was performed as described in Ingolia et al. (2009), with modifications from Stadler and Fire (2011).
Sequence analysis: Ribosome profiling
Ultra-high-throughput sequencing for both mRNA-seq and ribosome profiling experiments were performed using the Illumina platform. Reads were trimmed of all contiguous trailing A residues (introduced during library preparation by poly(A) polymerase), and resulting reads >20 nt were used for mapping. Reference sequences for mapping RPF and mRNA-seq reads consisted of the total set of predicted/verified coding sequences from the WormBase WS220 release with 18 nt of 5′ and 3′ genomic flanking sequence. For transcripts with multiple isoforms, a single isoform was selected arbitrarily. Reads were mapped to these reference sets using Bowtie v.0.12.2 (Langmead et al. 2009) reporting all reads that were perfect matches or contained a single mismatch to the reference.
Library sizes for all mRNA-seq and RPF libraries were normalized using the EdgeR (Robinson and Oshlack 2010) software package within R using the TMM normalization mode. Using normalized library sizes, the reads per kilobase of exon per million mapped reads (RPKM) (Mortazavi et al. 2008) were determined for each gene in each library. Ribosome loading was computed separately for each RPF library as the log2 ratio of normalized tag counts in that library and the average normalized tag count from all mRNA-seq libraries derived from the same developmental stage. Figures representing alternative normalization procedures are available in the Supplemental Material.
Positional difference scores for ribosome occupancy profiles were calculated using a sliding window approach. A single occupancy vector was generated for each gene at each stage by combining all biological replicates from a given stage and normalizing to the total coverage for the gene within that stage. A difference score for each window was calculated as (occupancystage1 – occupancystage2)/(occupancystage1 + occupancystage2), where occupancystage n represents the total occupancy (normalized read count) within the window in that stage. Using windows of 20 codons and steps of 10 codons, a distribution of difference scores was thus generated for each gene in a comparison between stages. The mean and median of these distributions were examined for miRNA targets and compared to transcripts of similar coverage levels (with similar coverage defined as ±0.40 reads per codon).
Coincidence analysis was used to determine how similar two ribosome occupancy profiles were. The coincidence statistic describes the probability that choosing a random sequencing read from each of two libraries will fall on the same position. More similar profiles will thus show a greater probability of coincidence, greater differences will thus result in a small probability of coincidence.
lin-14 heterozygote allele-specific mRNA measurements
lin-14 n536n539 (Ambros and Horvitz 1987) hermaphrodites were crossed with males harboring the integrated transgene hlh-8∷GFP on the X chromosome (Harfe et al. 1998), and GFP was used to mark hermaphrodite cross-progeny. Animals were hatched in the presence of food, and populations of five L1 (<3 h post-hatch) or five L4 animals were frozen in GITC buffer, followed by nucleic acid preparation as described in the single worm RT-PCR protocol in Epstein and Shakes (1995).
Reverse transcription of a lin-14 fragment surrounding the n539 point mutation was carried out using the primer AF-MS-51 (ATTTGCGCATTGCCTCGCGG). PCR was carried out with primers AF-MS-55 (AAGCGTGTCTTTGGACCACG) and AF-MS-56 (ATTTGTCCCAAAAGTCTTCC) and Phusion polymerase (NEB). Primers span a 45-bp intron, allowing clear separation on an agarose gel of RNA-derived product (165 bp) from products amplified from genomic DNA (210 bp). Because amplifications were carried out using relatively small amounts of starting cDNA, we sought to control for PCR jackpot effects by first carrying out 12 independent amplifications, combining the products in equal volumes, and performing several additional cycles of amplification.
Sequencing using Sanger sequencing chemistry was carried out by Elim Biopharmaceuticals using AF-MS-56 as the sequencing primer. To allow sequencing by the Illumina GAIIx system, several additional rounds of PCR were carried out using primers designed to add the necessary sequences for Illumina sequencing and 4-nt barcodes.
Immunoblot analysis
Immunoblots were carried out using standard procedures (Gallagher et al. 2011). Previously frozen worm pellets were normalized by weight. Each sample was resuspended in a volume four times its weight using 2× sample buffer (Laemmli Sample buffer from Bio-Rad, 1 mM PMSF, 1× Halt protease, and Phosphatase inhibitor from Thermo Scientific). Samples were boiled for 6 min, and loaded on a 10% acrylamide Criterion gel from Bio-Rad. For the mixing experiment, L1 sample was combined with L4 sample in the stated volume ratios. Proteins were transferred onto Milipore Immobilon FL membrane pre-wetted in 100% Methanol. Polyclonal antisera against 6HIS∷LIN-14(284–465) were a gift from Victor Ambros (Hristova et al. 2005). Polyclonal antisera against full-length HIS-tagged LIN-28 were a gift from Eric Moss (Seggerson et al. 2002). The anti-tubulin (beta-) E7 monoclonal antibody developed by M. Klymkowsky was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA. Blocking was carried out in 1× PBS containing 4% Milk, 1% BSA, 0.1% Tween, and 250 mM NaCl. Primary incubation was carried out overnight at 4°C in blocking buffer for LIN-14 and LIN-28. For tubulin, 0.5% BSA was substituted as the blocking agent. Secondary Cy3 Affinipure goat anti-mouse and Cy5 Affinipure goat anti-rabbit were from Jackson ImmunoResearch Laboratory Inc. Fluorescence imaging was carried out on a Typhoon Trio (Amersham Biosciences) and analyzed using ImageQuant v5.1.
Data access
All sequencing reads are available from the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra/) under accession number SRA055804.
Acknowledgments
We thank the following individuals for help and support: A. Sidow, Z. Weng, P. Lacroute, N. Ingolia, P. Sarnow, K. Wehner, G. Fuchs, V. Ambros, E. Moss, C. Krishna, J. Maniar, H. Zhang, S. Gu, P. Parameswaran, L. Gracey, C. Mello, A. Lamm, D. Wu, R. Ahrends, K. Kovary, and M. Teruel. This work was supported by grants to A.F. from NIGMS (RO1GM37706), and NIAID (U54065359). M.S. was supported by the Stanford Genome Training Program (T32-HD00044), Stanford Graduate Fellowship Program, and a National Science Foundation Graduate Research Fellowship.
Footnotes
-
↵3 Corresponding author
E-mail afire{at}stanford.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.136515.111.
- Received December 15, 2011.
- Accepted July 16, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








