
Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism
- Daniel Savic1,3,
- Honggang Ye2,
- Ivy Aneas1,
- Soo-Young Park2,
- Graeme I. Bell1,2 and
- Marcelo A. Nobrega1,3
- 1Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA;
- 2Department of Medicine, University of Chicago, Chicago, Ilinois 60637, USA
Abstract
Genome-wide association studies (GWAS) have consistently implicated noncoding variation within the TCF7L2 locus with type 2 diabetes (T2D) risk. While this locus represents the strongest genetic determinant for T2D risk in humans, it remains unclear how these noncoding variants affect disease etiology. To test the hypothesis that the T2D-associated interval harbors cis-regulatory elements controlling TCF7L2 expression, we conducted in vivo transgenic reporter assays to characterize the TCF7L2 regulatory landscape. We found that the 92-kb genomic interval associated with T2D harbors long-range enhancers regulating various aspects of the spatial–temporal expression patterns of TCF7L2, including expression in tissues involved in the control of glucose homeostasis. By selectively deleting this interval, we establish a critical role for these enhancers in robust TCF7L2 expression. To further determine whether variation in Tcf7l2 expression may lead to diabetes, we developed a Tcf7l2 copy-number allelic series in mice. We show that a null Tcf7l2 allele leads, in a dose-dependent manner, to lower glycemic profiles. Tcf7l2 null mice also display enhanced glucose tolerance coupled to significantly lowered insulin levels, suggesting that these mice are protected against T2D. Confirming these observations, transgenic mice harboring multiple Tcf7l2 copies and overexpressing this gene display reciprocal phenotypes, including glucose intolerance. These results directly demonstrate that Tcf7l2 plays a role in regulating glucose tolerance, suggesting that overexpression of this gene is associated with increased risk of T2D. These data highlight the role of enhancer elements as mediators of T2D risk in humans, strengthening the evidence that variation in cis-regulatory elements may be a paradigm for genetic predispositions to common disease.
Recent GWAS have uncovered a number of loci affecting risk of T2D (Voight et al. 2010). While some of these loci include genes known to play a role in glucose metabolism and diabetes pathogenesis (PPARG, KCNJ11) (Willson et al. 2001; Gloyn et al. 2006), others represent genomic regions with unknown functional roles in disease etiology. Among these, a set of single nucleotide polymorphisms (SNPs) on chromosome 10q25.2 shows strong and consistent association with T2D in virtually every population tested, constituting the greatest effect on risk identified to date, with a cumulative allelic odds ratio of 1.46 (Grant et al. 2006; Cauchi et al. 2007; Lyssenko 2008). These SNPs map to a 92-kb interval within TCF7L2, a gene encoding a transcription factor of the canonical Wnt signaling pathway known to have developmental roles in determining cell fate, survival, proliferation, and movement (Moon et al. 2004; Clevers 2006; MacDonald et al. 2009). Given the complexity of this pathway, establishing a definitive role for TCF7L2 in the etiology of T2D has been challenging (Pearson 2009).
While the TCF7L2 T2D-associated interval spans coding sequence, exonic variation has been excluded. This raises the question of what roles these noncoding variants play on disease etiology. Significant attention has been given to splicing as a potential disease mechanism as a result of the complex alternative transcripts generated by TCF7L2. Several reports have characterized the tissue-specific alternative splicing patterns of TCF7L2 (Osmark et al. 2009; Prokunina-Olsson et al. 2009a,b; Mondal et al. 2010; Prokunina-Olsson and Hall 2010), but none have provided a conclusive link between T2D-associated SNPs and splicing isoforms.
Recent studies have highlighted the importance of cis-regulatory variation in noncoding-associated intervals in affecting risk to common diseases, including colorectal and prostate cancers, blood cholesterol levels, and coronary artery disease (Musunuru et al. 2010; Pittman et al. 2010; Wasserman et al. 2010; Harismendy et al. 2011). Collectively, these studies immediately evoke a similar mechanism involved at the TCF7L2 T2D-associated region, implicating variation in long-range enhancers as the underlying causal link to disease risk. Recent reports corroborate this idea, showing that the SNP with the strongest degree of association in the interval (rs7903146) is within a region of open chromatin in pancreatic islets (Gaulton et al. 2010; Stitzel et al. 2010). Moreover, this effect is allele specific, with the risk allele (T) corresponding to open chromatin and increased enhancer activity compared with the nonrisk allele (C) (Gaulton et al. 2010; Stitzel et al. 2010). These data suggest that an increased risk of T2D may be associated with overexpression of Tcf7l2 in specific tissues.
To directly test the hypothesis that variation in Tcf7l2 expression levels is the functional link between the association of noncoding SNPs within this locus and T2D risk, we aimed (1) to address the potential role of cis-regulatory elements within the TCF7L2 association interval in T2D susceptibility, and (2) to establish through in vivo models the effect of altered Tcf7l2 expression on T2D phenotypes, thereby assessing a connection between cis-regulatory variation and disease risk.
We first characterized the long-range cis-regulatory landscape of this locus, demonstrating that the T2D-associated region harbors a wide variety of enhancers, including a subset that drives expression in tissues regulating glucose homeostasis. Importantly, we show that the enhancers located within the T2D-associated interval are critical for TCF7L2 expression. We further created an in vivo copy-number series of Tcf7l2 in mice (animals with 0, 1, 2, or 5 Tcf7l2 copies) and show a direct, dose-dependent role for altered Tcf7l2 expression on glucose metabolism. Our in vivo results predict that overexpression of TCF7L2 would lead to T2D through a lowered tolerance to glucose and further emphasize the potential role of alterations in cis-regulatory elements in regions of common disease risk.
Results
Bacterial artificial chromosome (BAC) enhancer trapping scan
We utilized four human BACs spanning 499.5 kb of sequence centered on TCF7L2 to scan broad expanses of noncoding DNA for regulatory potential in an in vivo mouse transgenic reporter assay (Fig. 1A). BACs spanning upstream (RP11-57H14), intragenic (RP11-466I19 and RP11-139K1) and downstream (RP11-357H24) sequences of TCF7L2 were converted into enhancer trapping clones by recombining a lacZ cassette into each BAC (Spitz et al. 2003; Wasserman et al. 2010). Both of the intragenic BACs spanned the entire T2D-associated interval, including SNP rs7903146 (T/C), the variant exhibiting the strongest association to T2D (Fig. 1A, black asterisk). By genotyping SNP rs7903146, we determined that both BACs harbored the protective, derived C allele. We generated multiple independent mouse transgenic lines for each genetically modified BAC and assayed beta-galactosidase activity at various embryonic and postnatal stages of development.
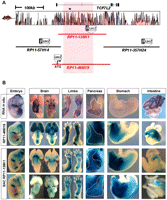
The regulatory landscape of TCF7L2. (A) The location of enhancer-trapping human BACs (RP11-57H14, RP11-466I19, RP11-139K1, and RP11-357H24) spanning 499.5 kb at the TCF7L2 locus. Noncoding sequence conservation between human and mouse is displayed as red peaks. The T2D-associated interval is highlighted in red. A black asterisk marks the location of SNP rs7903146. (B) The endogenous expression of Tcf7l2 is shown in the top row, while enhancer-trapped BAC RP11-466I19 and two independent BAC RP11-139K1 transgenic lines are shown in the middle and bottom rows, respectively. Whole-mount images at embryonic day 12.5 (E12.5) are shown in the first column. Images of brain (dorsal and ventral views), limb (fore- and hindlimbs), pancreas (outlined with dotted white line), stomach, and intestine at E15.5 are shown in the adjacent columns (left to right). Enhancer-trapped BACs RP11-466I19 and RP11-139K1 show overlap with endogenous Tcf7l2 expression. Enhancer activity is present in the brain, fore- and hindlimbs, pancreas, pyloric stomach, and lower gastrointestinal tract.
We found that BACs containing sequences outside of the 92-kb T2D-associated interval generated limited enhancer activity (Supplemental Fig. S1). Sequences upstream of TCF7L2 contained enhancer activity within the forebrain and olfactory epithelium, while sequences downstream from TCF7L2 exhibited expression within the axial skeleton. Collectively, this ∼300-kb region recapitulated only a fraction of the endogenous Tcf7l2 expression profile.
In contrast, two different BACs spanning the T2D-association interval gave highly reproducible beta-galactosidase expression patterns that mimicked the majority of the endogenous Tcf7l2 expression, including enhancers driving expression within the diencephalon, limbs, pancreas, and intestine, each of which were maintained from embryonic day 12.5 into adulthood (Fig. 1B; Supplemental Fig. S2). While both BACs exhibited largely concordant expression profiles, we did observe variation in brain expression. This may be attributed to differences in the position of integration of the reporter gene, which was recombineered in-frame with the TCF7L2 translation start site in BAC RP11-466I19, but randomly integrated in BAC RP11-139K1. Alternatively, the difference in sequence coverage between both BACs could also result in the slightly different global expression pattern obtained between the BACs. Interestingly, these BACs contained human-specific regulatory activity within the pyloric stomach and brain. From closer examinations of beta-galactosidase staining, we noted that embryonic intestinal expression was restricted to the distal regions of the gut, but expression in postnatal mice was visible in both the colon and upper gastrointestinal tract (Supplemental Fig. S2). Histological sections confirmed beta-galactosidase expression within the thalamus and hypothalamus of the brain, bone and skeletal muscles of the limbs, and the exocrine pancreas (Supplemental Fig. S3). Using immunohistochemistry, we further identified pancreatic expression within islets in neonatal mice (Supplemental Fig. S4). Surprisingly, we failed to identify pancreatic islet staining in adult mice. Supporting this observation, the inactivation of canonical Wnt signaling in pancreatic islets of adult mice was previously reported (Krutzfeldt and Stoffel 2010). Importantly, this long-range enhancer screen revealed that most of the cis-regulation of TCF7L2 arises from enhancers mapping to the intragenic region encompassing the entire T2D-associated interval.
Defining the regulatory importance of the T2D-association interval
To directly test the impact of the 92-kb T2D-associated interval on TCF7L2 expression, we selectively deleted this region within the enhancer-trapped BAC RP11-466I19 (with lacZ integrated in the translation start site of TCF7L2), and generated multiple transgenic mouse lines harboring this engineered BAC (Fig. 2A). The deletion of the T2D-associated segment resulted in a dramatic reduction of beta-galactosidase expression in most tissues. All four transgenic lines harboring the deletion exhibited either a significant reduction or complete ablation of beta-galactosidase staining within the pancreas, intestine, stomach, and appendicular skeleton (Fig. 2B). Brain expression was unaltered in deleted BACs, suggesting regulatory sequences governing brain expression lie outside of the T2D-associated region. However, the presence of redundant brain enhancers within the association interval cannot be excluded. Collectively, these results show that the T2D-association interval contains regulatory elements critical for TCF7L2 expression in multiple tissues that play an important role in glucose homeostasis, such as the pancreas, intestine, and skeleton (Lee et al. 2007). Moreover, these findings support the hypothesis that genetic variation within cis-regulatory elements at the 92-kb segment associated with T2D could potentially alter TCF7L2 expression in critical tissues and lead to an increased risk of T2D.
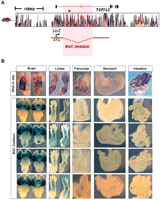
Regulatory importance of T2D-associated interval. (A) The location of enhancer-trapping human BAC RP11-466I19 containing a deletion of the association interval (BAC Deletion) at the TCF7L2 locus. Noncoding sequence conservation between human and mouse is displayed as red peaks (VISTA genome browser). The type 2 diabetes (T2D)–associated interval is highlighted in red. A black asterisk marks the location of SNP rs7903146. (B) The endogenous expression of Tcf7l2 is shown in the top row. Four independent transgenic lines of mice harboring the enhancer-trapped BAC Deletion are shown below. Images of brain (dorsal and ventral views), limb (fore- and hindlimbs), pancreas (outlined with dotted white line), stomach, and intestine at E15.5 are shown (left to right). Deletion of T2D-associated interval results in a complete ablation of enhancer activity in the pyloric stomach along with a reduction of enhancer activity in both fore- and hindlimbs, pancreas, and intestine in all four transgenic lines.
A dosage-dependent impact of Tcf7l2 on blood glucose levels in mice
To test whether alteration in Tcf7l2 expression has a phenotypic effect on glucose metabolism and T2D susceptibility in vivo, we generated an allelic series of Tcf7l2 copy number in mice. To ablate Tcf7l2, we generated null alleles using zinc finger nuclease (ZFN) technology (Geurts et al. 2009) targeted to the constitutive exon 11 of the murine Tcf7l2 gene, corresponding to the High Mobility Group (HMG) box DNA-binding domain of the protein. The use of this targeting strategy would ultimately lower the levels of functional TCF7L2 protein, thereby mimicking a loss-of-function mutation in a cis-regulatory element. Indeed, disruption of this same region was previously utilized and reported to be successful in ablating murine TCF7L2 function (Korinek et al. 1998). Using ZFNs, we obtained three independent mouse lines harboring frameshift-inducing deletions of 1-bp to 13-bp on the DNA-binding domain and premature stop codons (Fig. 3A; Supplemental Fig. S5).
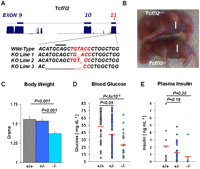
Generation of Tcf7l2 null mice using zinc finger nuclease technology. (A) Mouse Tcf7l2 gene exons 9, 10, and 11 are shown. The 6-bp of sequence targeted in exon 11 by zinc finger nucleases (ZFNs) is shown below in red along with flanking sequences. A horizontal line marks a codon. Deletions from the three knockout lines generated are shown below the wild-type sequence. (B) Images of Tcf7l2+/+ and Tcf7l2−/− mice that show a lack of milk within the stomach (white arrows). (C) Neonatal body weights for wild-type (+/+, n = 37), heterozygous null (+/−, n = 81), and homozygous null (−/−, n = 25) mice. (D) Scatter plots of neonatal blood glucose measurements for wild-type (+/+, n = 36), heterozygous null (+/−, n = 83), and homozygous null (−/−, n = 19) mice. Red bars mark the mean blood glucose level. (E) Plasma insulin values for wild-type (+/+, n = 9), heterozygous null (+/−, n = 24), and homozygous null (−/−, n = 6) animals. Red bars mark the mean insulin level.
Tcf7l2 homozygote null mice (Tcf7l2−/−) were born in Mendelian expectations but died perinatally, within ∼24 h of birth, consistent with the knockout mice engineered by Korinek et al. (1998). We further determined that Tcf7l2−/− mice lacked milk in the stomach, when wild-type and heterozygous null (Tcf7l2+/−) littermates had already successfully nursed (Fig. 3B), and had significantly lower body weights than wild-type and heterozygous knockout littermates (1.37 g of Tcf7l2−/− vs. 1.53 g of Tcf7l2+/− and 1.55 g of Tcf7l2+/+, P < 0.001) (Fig. 3C). Importantly, these phenotypes are analogous to those observed in a recent report of an independent TCF7L2 knock-out mouse model, where exon 1 of Tcf7l2 was disrupted (Angus-Hill et al. 2011).
To directly assess the impact of TCF7L2 on glucose homeostasis, we measured glucose (Fig. 3D) and insulin (Fig. 3E) levels in neonatal mice fasted for 4–6 h. We observed a dosage-dependent effect of Tcf7l2 on neonatal blood glucose levels. Tcf7l2−/− mice displayed severe hypoglycemia at birth (27.9 mg/dL), while Tcf7l2+/− mice maintained intermediate glucose levels (44.4 mg/dL) compared with wild-type littermates (53.8 mg/dL). While insulin levels exhibited a pattern that mirrored blood glucose levels, the differences between the groups were not significant.
Tcf7l2 heterozygous null mice are protected from diabetes
The early mortality of Tcf7l2−/− mice hampered studies of glucose and energy metabolism. We therefore focused on phenotyping heterozygous Tcf7l2+/− mice. Tcf7l2+/− mice displayed a consistent 10%–15% lower body weight compared with wild-type littermates (Fig. 4A). Dual-Energy X-ray Absorptiometry (DEXA) scans determined that this reduced body weight is due to both lower lean body and fat mass (Fig. 4B). Importantly, female Tcf7l2+/− mice exhibited these identical phenotypes (Supplemental Fig. S6). Despite their smaller size, Tcf7l2+/− mice are hyperphagic (Fig. 4C) and display reduced motor activity (Supplemental Fig. S7), while maintaining largely consistent energy expenditure compared with wild-type littermates (Supplemental Fig. S7).
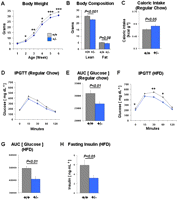
Characterization of Tcf7l2+/− mice. (A) Growth curves for wild-type (gray, n = 24) and heterozygous null (blue, n = 26) mice from 1 to 6 wk of age. (B) Lean and fat body mass in wild-type (+/+, n = 12) and heterozygous null (+/−, n = 13) mice. (C) Caloric intake in wild-type (+/+, n = 9) and heterozygous null (+/−, n = 10) mice. (D) Intraperitoneal glucose tolerance test (IPGTT) curves of wild-type (gray, n = 9) and heterozygous null (blue, n = 10) mice on a regular chow diet. (E) Area under the curve (AUC) of the IPGTT plot from D. (F) IPGTT curves of wild-type (gray, n = 11) and heterozygous null (blue, n = 12) mice after 10 wk on a high-fat diet (HFD). (G) Area under the curve (AUC) of the IPGTT plot from F. (H) Fasting plasma insulin levels in wild-type (+/+, n = 11) and heterozygous null mice (+/−, n = 12) after 10–12 wk on a HFD. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001.
Importantly, Tcf7l2+/− mice displayed an enhanced glucose tolerance, both on a regular chow diet and after a high-fat diet (HFD) regimen for 10 wk (Fig. 4D–G). Further, the Tcf7l2+/− mice on a HFD had significantly lower fasting (4 h) glucose and insulin levels than wild-type littermates (Fig. 4F,H).
Humanized Tcf7l2 overexpressing BAC transgenic mice are prone to diabetes
Having established that abrogation of Tcf7l2 leads to enhanced glucose tolerance, we next sought to determine the impact of increased expression of this gene on glucose and energy metabolism. To assess this, we engineered mice carrying extra copies of Tcf7l2, utilizing a bacterial artificial chromosome (BAC) recombineering strategy. A full-length mouse Tcf7l2 cDNA was recombined into the TCF7L2 translation start site within a 195-kb human BAC (RP11-466I19) utilized for our enhancer screen that spans the entire 92-kb T2D-association interval (Fig. 5A). With this strategy, we ensured (1) that the transgene is expressed in a pattern that closely mimics the endogenous expression profile, and (2) that the transgene is expressed in a human-like fashion, minimizing any potential human–mouse expression differences. This is important in the light that SNP rs7903146 resides within a repetitive DNA sequence present in humans but lacking in mice. We generated humanized Tcf7l2 BAC transgenic mice and determined, by quantitative PCR, that these mice harbor three copies of the BAC construct (Supplemental Fig. S8), resulting in a total of five Tcf7l2 copies in hemizygous mice. Quantitative RT–PCR (Fig. 5B) and RNA in situ hybridization (Fig. 5C) further demonstrated the increased expression of Tcf7l2 in multiple tissues, including brain, stomach, intestine, and pancreas. Importantly, these humanized mice harbored regulatory activities that were inconsistent with the endogenous expression in the mouse, such as in the forebrain (Fig. 5C), confirming that human-specific regulatory activity is driving mouse Tcf7l2 cDNA expression.
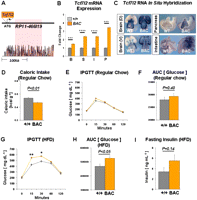
Characterization of Tcf7l2 BAC transgenic mice. (A) Schematic of human BAC RP11-466I19 recombined with a full-length mouse Tcf7l2 cDNA. (Below) Noncoding sequence conservation between human and mouse is marked by red peaks (VISTA genome browser). The T2D-associated interval is highlighted in red. (B) Tcf7l2 mRNA expression from wild-type (gray, n = 4) and Tcf7l2 BAC transgenic (orange, n = 4) mice in brain (B), stomach (S), intestine (I), and pancreas (P). (C) RNA in situ hybridizations of Tcf7l2 BAC transgenic (BAC) and wild-type (WT) tissues at embryonic day 15.5 (E15.5) show overexpression of Tcf7l2 in the brain, pancreas, and intestine in Tcf7l2 BAC transgenic mice. (D) Caloric intake in wild-type (+/+, n = 7) and Tcf7l2 BAC transgenic (BAC, n = 5) mice. (E) Intraperitoneal glucose tolerance test (IPGTT) curves of wild-type (gray, n = 13) and Tcf7l2 BAC transgenic (orange, n = 9) mice on a regular chow diet. (F) Area under the curve (AUC) of the IPGTT plot from E. (G) IPGTT curves of wild-type (gray, n = 8) and Tcf7l2 BAC transgenic (orange, n = 6) mice after 10 wk on a high-fat diet (HFD). (H) Area under the curve (AUC) of the IPGTT plot from G. (I) Fasting plasma insulin levels in wild-type (gray, n = 10) and Tcf7l2 BAC transgenic (orange, n = 8) mice after 10–12 wk on a HFD. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001.
Growth curves and DEXA scans determined Tcf7l2 BAC transgenic mice did not show differences in body mass or composition respectively (Supplemental Fig. S8). However, Tcf7l2 BAC transgenic mice did display a host of reciprocal phenotypes compared with Tcf7l2 null mice, including hypophagia (Fig. 5D), while further maintaining consistent energy expenditure (Supplemental Fig. S8). Importantly, we found that overexpression of Tcf7l2 in our BAC transgenic mice lead to significant glucose intolerance, after a HFD regimen for 10 wk (Fig. 5G,H). Further, the Tcf7l2 BAC transgenic mice on a HFD showed a trend for an increase in fasting (4 h) insulin levels compared with wild-type littermates (Fig. 5I). Together, our results demonstrate a direct, dosage-dependent effect of Tcf7l2 on glucose homeostasis, with an increase in copy number leading to progressive impairment of glucose tolerance.
Discussion
Our study provides a functional in vivo connection between TCF7L2 and the etiology of type 2 diabetes, supporting the notion that regulatory variation within Tcf7l2 that leads to an overexpression of this gene may underlie the increased risk to T2D.
We characterized the cis-regulatory map of the TCF7L2 locus. Using an in vivo long-range BAC enhancer screen strategy, we identified the intronic regions of TCF7L2 as harboring the majority of the endogenous Tcf7l2 expression. Moreover, through the selective deletion of the T2D-associated interval in our enhancer-trapped human BAC, we further demonstrate that the enhancers situated within the association interval were critical for robust TCF7L2 expression in tissues with known roles in regulating glucose metabolism.
In support of a role for cis-regulatory variation in T2D susceptibility, our in vivo mouse models displayed defects in glucose homeostasis through alterations in Tcf7l2 expression. To our knowledge, this is the first in vivo model demonstrating a role for TCF7L2 in glucose homeostasis and T2D susceptibility. We show that a dose-dependent increase in TCF7L2 leads to progressively weaker glucose tolerance in vivo. These data are consistent with studies in humans showing that the T2D-risk allele is associated with higher TCF7L2 mRNA levels, is in an open chromatin conformation, and has increased enhancer activity in an in vitro assay (Lyssenko et al. 2007; Gaulton et al. 2010; Stitzel et al. 2010). Our in vivo results would therefore predict that an overexpression of Tcf7l2 would lead to an increased susceptibility to T2D. Moreover, these data solidify TCF7L2 as the target gene emerging from the multiple association studies with T2D.
While our Tcf7l2 overexpression mice harbor transgenic BACs containing the T2D protective allele at SNP rs7903146, we effectively achieved overexpression of Tcf7l2, which is presumed to be the effect of the risk allele at rs7903146 (Gaulton et al. 2010; Stitzel et al. 2010) and, potentially, elsewhere in the linkage disequilibrium block. An ideal design, in which allele-specific expression differences could be phenotypically measured, is difficult to achieve using mouse transgenesis, due to both position and copy-number variegation between independent lines. We believe that such experiments will be more efficiently carried out in cell-based assays, such as those reported by Gaulton et al. (2010) and Stitzel et al. (2010).
The use of a mouse model in our study allows for a diverse tool kit for genetic manipulation, which we exploited, as well as a comprehensive analysis within an in vivo setting. This latter aspect may account for the conflicting observations reported by in vitro studies (Loder et al. 2008; Shu et al. 2008) as these experiments were restricted to pancreatic islets, and consequently did not capture tissue interactions and TCF7L2 function outside of the pancreas. The use of a rodent species for following-up of phenotypic effects observed in humans may also generate difficulties in data interpretation. One chief concern is the degree of evolutionary conservation in the regulation of biological processes between humans and mice. Notably, our method tried to circumvent this drawback by designing a humanized mouse containing human regulatory sequences. Along the same vein, we were able to utilize the entire human association interval, including sequences spanning SNP rs7903146, which lies within a repetitive element that is absent in mice.
The regulatory signature identified by our scan establishes the potential role of cis-regulatory variation at the TCF7L2 association interval in T2D risk. Indeed, experimental follow-ups to GWAS have systematically found regulatory activity in regions of disease association (Musunuru et al. 2010; Pittman et al. 2010; Wasserman et al. 2010), suggesting that this may be a paradigm for common diseases. Further, while studies on the role of TCF7L2 in T2D susceptibility have largely focused on pancreatic islets (Loos et al. 2007; Liu and Habener 2008; Loder et al. 2008; Shu et al. 2008, 2009; da Silva Xavier et al. 2009; Khalooghi et al. 2009), our unbiased approach localized cis-regulatory activity for a wide array of tissues involved in glucose homeostasis to the associated interval, suggesting that the role of TCF7L2 in the pathogenesis of T2D may not be solely limited to the pancreas. To that end, we did not identify pancreatic islet expression in adult mice, and the same SNP rs7903146 associated with T2D has also been associated with an increased risk of colorectal cancer (Folsom et al. 2008), independent of diabetes.
As a follow up to these results, an unbiased fine-mapping enhancer screen can be used within the T2D-associated interval by interrogating smaller intervals for regulatory potential using reporter constructs that can be tested in vivo or in vitro. While the association interval spans a broad expanse of sequence, enhancer fine-mapping can be aided through the use of various publicly available data sets (such as ENCODE) for transcription-factor binding, chromatin state, and histone modifications to identify sequences that may be predicted to harbor regulatory activity.
From a common disease standpoint, variation within cis-regulatory elements is a means of compartmentalizing phenotypic effects, wherein the biological disruption is limited to a spatial–temporal subset of the global function of the gene and would, therefore, be predicted to result in a less detrimental effect. Our findings that the strongest genetic signal associated with common forms of diabetes in humans is connected to regulatory variation bring this disease to the rapidly increasing hall of complex disorders that have regulatory mutations as their underpinnings.
Methods
BAC recombineering
BACs RP11-57H14, RP11-139K1, and RP11-357H24 (Children's Hospital Oakland Research) were modified by an in vitro random transposition of a vector containing a Tn7 transposon site flanking a beta-globin minimal promoter driving lacZ and a kanamycin resistance gene using the Tn7 Genome Priming System (New England Biolabs). A total of 20 ng of vector DNA, 25 ng of BAC DNA, GPS buffer, and a TnsABC transposase were mixed followed by a 10-min incubation at 30°C, addition of Start Solution and subsequent incubation at 37°C for 1 h, and a final heat inactivation at 75°C for 10 min. To minimize salt, a 1-h dialysis using Millipore MF-Membrane Filters (0.025 μm) was performed in Embryo water (Sigma). A total of 2 μL of transposition reaction was electroporated into 20 μL of electro-competent DH10B cells (Invitrogen). Cells were plated on Luria-Bertani (LB) agar containing 20 μg/mL of kanamycin and 12.5 μg/mL of chloramphenicol and grown overnight at 37°C. Colonies were grown at 37°C in LB cultures containing the above concentration of antibiotics. DNA was extracted using a Qiagen Miniprep Kit (Qiagen). Positive clones were initially identified through PCR. Restriction enzyme fingerprinting was conducted to determine the location of integration.
BAC RP11-466I19 was modified by a lacZ reporter gene using the RED/ET Recombination Kit (Gene Bridges). RED/ET plasmid was electroporated into a BAC RP11-466I19 containing bacterial culture (chloramphenicol-resistant) and plated for an overnight incubation on LB agar containing 3 μg/mL of tetracycline and 12.5 μg/mL of chloramphenicol at 30°C in the dark. Subsequent colonies were grown into LB cultures containing the above concentration of antibiotics at 30°C in the dark. A total of 30 μL of overnight culture was inoculated into 1.4 mL of LB culture containing the above concentration of antibiotics in a 2-mL microcentrifuge tube with the lid punctured. Cultures were grown at 30°C in the dark until OD600 ∼0.3. Then 50 μL of 10% L-arabinose was added and the cultures were allowed to grow at 37°C for 1 h. Primers, modified by 50 nucleotides of overhanging sequence with homology with the TCF7L2 start site, were designed to amplify a lacZ and ampicillin-resistance gene. PCR reactions were purified using the QIAquick PCR Purification Kit (Qiagen) and dialyzed for 1 h using a Millipore MF-Membrane Filters (0.025 μm) in Embryo water (Sigma). A total of 200 ng of purified PCR product was electroporated into BAC containing RED/ET plasmid cultures with L-arabinose and plated on LB agar containing 12.5 μg/mL of chloramphenicol and 50 μg/mL of ampicillin and grown overnight at 37°C. Resulting colonies were analyzed for accurate recombination using restriction enzyme fingerprinting and by sequencing recombination endpoints.
The deletion of the association interval within enhancer-trapped BAC RP11-466I19 was done using the RED/ET Recombination Kit through the use of primers designed to amplify a kanamycin resistance gene that additionally contained 50 nucleotides of overhanging sequence that flanked the association interval. Positive clones were selected for growth on 20 μg/mL of kanamycin and 50 μg/mL of ampicillin and assayed for deletions using restriction enzyme fingerprinting and sequencing of deletion breakpoints.
Preparation of DNA for microinjection
BAC DNA was purified using the Nucleobond BAC 100 Kit (Macherey-Nagel) and resuspended in Embryo water (Sigma). Thirty micrograms of BACs RP11-466I19, RP11-139K1, and RP11-357H24 were linearized by an overnight Asc1 restriction digestion at 37°C. The linearized BAC was diluted 1:20 in 1× microinjection buffer (10 mM Tris at pH 7.5; 0.1 mM EDTA) and run on a Sepharose column to isolate BAC DNA in 6–500-μL fractions. Fraction concentrations were determined and a pulsed-field gel using 1% agarose was run on 60-μL aliquots from each fraction to determine the integrity of the BACs. BACs were then diluted to 4 ng/μL using 1× microinjection buffer.
BAC RP11-57H14 was injected in its circular form by an initial resuspension in 1× microinjection buffer after purification using the Nucleobond BAC100 Kit (Macherey-Nagel). After concentration was determined, 1× microinjection buffer was used to dilute the BAC to 4 ng/μL. A 1% agarose gel was run to detect the integrity of the circular BAC.
Purified BACs were used for pronuclear injections of CD-1 fertilized oocytes (Charles River) in accordance with standard protocols approved by the University of Chicago.
Mouse in vivo transgenic reporter assays
All animals were anesthetized using carbon dioxide followed by cervical dislocation. For prenatal analyses, embryos were harvested at E12.5, E15.5, and E17.5. Postnatal studies were conducted on mice 3-wk of age. Following harvesting and dissection, embryos and tissues were placed into cold 100-mM phosphate buffer at pH 7.3 (PBS), followed by 1 h of incubation with 4% paraformaldehyde at 4°C. Tissues were then washed with 1× PBS and further washed two additional times for 20 min using LacZ wash buffer (2 mM MgCl2; 0.01% deoxycholate; 0.02% NP-40; 100 mM phosphate buffer at pH 7.3), and stained for 16–20 h at room temperature with LacZ staining solution (1 mg/mL X-gal; 4 mM potassium ferrocyanide; 4 mM potassium ferricyanide; 20 mM Tris at pH 7.5 in wash buffer). After staining, embryos and tissues were rinsed five times in PBS and post-fixed in 4% paraformaldehyde indefinitely at 4°C. Images were taken using a Leica MZ 16 F imaging system.
Generation of knockout mouse lines
Zinc finger nucleases were designed to target exon 11 (using alternative exon 4 in exon numbering) of the murine Tcf7l2 gene. Zinc finger nuclease RNA was prepared as previously described (Geurts et al. 2009) and injected in fertilized CD-1 (Charles River) mouse oocytes. Knockout mice were identified using the SURVEYOR nuclease kit (Transgenomic). DNA was isolated from tail samples, amplified through PCR using primers flanking the ZFN targeted exonic region (forward 5′-AGCAAAGCATCGAGTCTCATTT-3′, reverse 5′-AAGCAGGTAGTAAGCCATCCTCT-3′), purified using the QIAquick PCR Purification Kit (Qiagen), and digested using the SURVEYOR assay. DNA was run on a 3% agarose gel in 0.5× TBE and visualized to identify knockouts. Sequencing was additionally preformed as a second verification of deletions. All three knockout lines were used for neonatal studies, while two knockout lines were utilized for adult phenotyping.
Generation of overexpressing transgene mouse lines
A full-length mouse Tcf7l2 (Open Biosystems) was recombined into the human TCF7L2 start site in BAC RP11-466I19 (Children's Hospital Oakland Research) using the RED/ET Recombination Kit (Gene Bridges). A detailed procedure is described above. BAC copy number was determined as previously described (Chandler et al. 2007). Male mice were used for phenotyping.
Quantitative reverse-transcription PCR
Neonates (1–2 d of age) were decapitated, and brain, pancreas, stomach, and intestines were dissected out in DEPC-treated 1× PBS. Tissues were flash-frozen in liquid nitrogen and stored at −80°C. RNA was extracted from wild-type and BAC-overexpressing tissues using Tri-Reagent (Sigma). cDNA was generated using Superscript II reverse transcriptase (Invitrogen). Tcf7l2 exon primers and HPRT housekeeping gene primers were used to quantify Tcf7l2 mRNA levels. RNA samples with no reverse transcriptase added were used in conjunction in RT–qPCR reactions to test for genomic DNA contamination.
RNA in situ hybridization
RNA in situ hybridization analysis using digoxigenin-labeled Tcf7l2 antisense riboprobes was performed using standard protocols. The probes were generated from a full-length mouse Tcf7l2 cDNA clone (Open Biosystems). Staining was performed for 2–4 h, followed by a transfer of tissues to 10% buffered formalin phosphate at 4°C indefinitely. Images were taken using a Leica MZ 16 F imaging system. Tissues from transgenic BAC mice and wild-type littermates were processed in parallel.
Neonatal in vivo metabolic testing
Neonatal mice were weighed prior to blood glucose testing. Neonates were decapitated and an AccuChek Aviva glucometer (Roche) was used to measure blood glucose. Blood glucose values below 10 mg/dL could not be determined. A total of 10 mg/dL was used for neonate blood glucose levels that fell below this threshold. Stomach weights using wild-type and heterozygous animals confirmed that the differences seen in body weights could not be attributed to the absence of milk in homozygous null animals. Blood for insulin levels was taken from decapitated neonates and collected in EDTA-coated microcentrifuge tubes and placed on ice. Blood was spun at 13,500 RPM for 15 min and the plasma was stored at −80°C. A rat/mouse insulin ELISA kit (Millipore) was used to quantify plasma insulin concentrations.
Adult in vivo metabolic testing
Mice had access to food and water. Both a regular chow (Harlan Teklad) and a high-fat diet (55% fat, Harlan Teklad) were used. Growth curve analyses were conducted by weighting litters on a weekly basis. Weaned mice were housed up to five per cage. Weighing was concluded at 6 wk of age. To test for energy homeostasis phenotypes, mice were housed in metabolic cages (TSE Systems) for a total of 7 d. Measurements for days 6 and 7 were used in the final analysis to ensure that the animals were fully acclimated. Dual-Energy X-ray Absorptiometry (DEXA) scans were preformed using a Lunar PIXImus II (GE Medical Systems). Prior to scanning, animals were anesthetized with ketamine, and their body length was determined. Blood glucose measurements from IPGTTs were taken on mice that fasted for 4 h. Mice were intraperitoneal injected with 2 mg/kg (20%) of dextrose, and their blood glucose taken using an AccuChek Aviva glucometer (Roche). Blood for insulin levels was taken from tail samples and collected in EDTA-coated microcentrifuge tubes and placed on ice. Blood was spun down and the plasma was stored at −80°C. A rat/mouse insulin ELISA kit (Millipore) was used to quantify plasma insulin concentrations. IPGTT measurements were carried out at 6 wk of age (knockout) and at 7–8 (transgenic BAC) wk of age under a regular chow diet, as well as at 17–18 wk of age after 10 wk on a high-fat diet. Plasma insulin was taken on mice after 10–12 wk on a high-fat diet.
Statistical analyses
Data are shown as mean ± SEM). An unpaired two-sided Student's t-test was utilized to test for significance, while a Bonferroni correction was used to correct for multiple testing.
Acknowledgments
We thank François Spitz for the Tn7-lacZ plasmid; Kenneth Polonsky for critical reading of the manuscript; Clay Semenkovich and David Ehrmann for their thoughtful comments; the Kover Family Foundation for their gift; and the Diabetes Research and Training Center (DRTC) at the University of Chicago. This work was supported by NIH grants DK-078871 and HG004428 (M.A.N.), DK-20595 (G.I.B.), and T32 GM007197.
Footnotes
-
↵3 Corresponding authors.
E-mail nobrega{at}uchicago.edu.
E-mail dsavic{at}uchicago.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.123745.111.
- Received March 25, 2011.
- Accepted June 9, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








