
Dioxin receptor and SLUG transcription factors regulate the insulator activity of B1 SINE retrotransposons via an RNA polymerase switch
- Angel Carlos Román1,
- Francisco J. González-Rico1,
- Eduardo Moltó2,3,
- Henar Hernando4,
- Ana Neto5,
- Cristina Vicente-Garcia2,3,
- Esteban Ballestar4,
- José L. Gómez-Skarmeta5,
- Jana Vavrova-Anderson6,
- Robert J. White6,7,
- Lluís Montoliu2,3 and
- Pedro M. Fernández-Salguero1,8
- 1 Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias, Universidad de Extremadura, 06071 Badajoz, Spain;
- 2 Centro Nacional de Biotecnología (CNB), Consejo Superior de Investigaciones Científicas (CSIC), Department of Molecular and Cellular Biology, Campus de Cantoblanco, C/Darwin 3, 28049 Madrid, Spain;
- 3 Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII, Madrid, Spain;
- 4 Chromatin and Disease Group, Cancer Epigenetics and Biology Programme, Bellvitge Biomedical Research Institute (IDIBELL), Barcelona 08907, Spain;
- 5 Centro Andaluz de Biología del Desarrollo, CSIC-Universidad Pablo de Olavide, 41013 Sevilla, Spain;
- 6 College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow G12 8QQ, United Kingdom;
- 7 Beatson Institute for Cancer Research, Glasgow, G61 1BD, United Kingdom
Abstract
Complex genomes utilize insulators and boundary elements to help define spatial and temporal gene expression patterns. We report that a genome-wide B1 SINE (Short Interspersed Nuclear Element) retrotransposon (B1-X35S) has potent intrinsic insulator activity in cultured cells and live animals. This insulation is mediated by binding of the transcription factors dioxin receptor (AHR) and SLUG (SNAI2) to consensus elements present in the SINE. Transcription of B1-X35S is required for insulation. While basal insulator activity is maintained by RNA polymerase (Pol) III transcription, AHR-induced insulation involves release of Pol III and engagement of Pol II transcription on the same strand. B1-X35S insulation is also associated with enrichment of heterochromatin marks H3K9me3 and H3K27me3 downstream of B1-X35S, an effect that varies with cell type. B1-X35S binds parylated CTCF and, consistent with a chromatin barrier activity, its positioning between two adjacent genes correlates with their differential expression in mouse tissues. Hence, B1 SINE retrotransposons represent genome-wide insulators activated by transcription factors that respond to developmental, oncogenic, or toxicological stimuli.
Mammalian genes are often clustered at chromosomal locations sharing common cis-regulatory elements (Gerasimova and Corces 2001; Bushey et al. 2008; Moltó et al. 2009). Such organization in expression domains poses the problem of misregulation due to inappropriate action of enhancers (Bell et al. 2001). To cope with this, insulators have evolved to delineate functionally independent chromosomal regions (Bell et al. 2001; Gerasimova and Corces 2001; Ohlsson et al. 2001; Mukhopadhyay et al. 2004) that might be exploited to improve the success of gene-therapy protocols and transgenesis experiments by modulating gene expression (Giraldo et al. 2003; Recillas-Targa et al. 2004; Moltó et al. 2009).
It is known that transposable elements such as ERV1, ERVK, MIR1, LTR10, and MER61 contain binding sites for transcription factors (Wang et al. 2007; Bourque et al. 2008; Kuwabara et al. 2009; Kunarso et al. 2010). Moreover, a B2 SINE retrotransposon has been shown to insulate expression domains during murine organogenesis (Lunyak et al. 2007). Therefore, it is of great interest to determine whether retrotransposons interact with specific transcription factors to define genome-wide expression profiles and whether they require the epigenetic machinery. In plants, epigenetics appear to play an important role in transposon-mediated control of gene expression (Lippman et al. 2004). However, in mouse and humans no direct link has yet been established between transposon epigenetics and control of gene expression (Aravin et al. 2007).
A SINE B1-X35S retrotransposon was found in over 14,000 instances in the mouse genome that contains a dioxin receptor (AHR) binding site (XRE) (common to canonical B1 elements) and a SLUG (SNAI2) site for recruitment of the zinc-finger transcription factor SLUG. Sequence conservation analysis revealed that B1-X35S is a new SINE-B1subfamily differing from canonical elements by the presence of the SLUG site (Roman et al. 2008). We previously observed that B1-X35S is enriched in promoter regions (1398 genes) and that AHR and SLUG binding represses transcription of downstream genes. However, the mechanism by which B1-X35S-containing genes are repressed by AHR and SLUG remained unsolved. Here, we demonstrate that B1-X35S SINE can also function as an insulator. This insulation mechanism is complex, involving transcription by Pol III and Pol II, accumulation of heterochromatic marks at downstream regions, and binding of CTCF. AHR is a well-conserved and ubiquitously expressed basic-helix-loop-helix (bHLH) transcription factor with a prominent role in cell physiology and homeostasis and in the response to environmental toxins (Furness et al. 2007; Gomez-Duran et al. 2009). SLUG has a critical role in epithelial-to-mesenchymal transitions that occur during development and in certain pathological conditions, such as tumor metastasis (Thiery et al. 2009; Martinez-Estrada et al. 2010). B1-X35S SINEs repress gene expression in vivo as the result of AHR and SLUG binding to their cognate consensus elements (Roman et al. 2008). The abundance of B1-X35S in the mouse genome suggests a widespread impact on gene expression. This may be important during normal development as well as in pathological conditions.
Results
B1-X35S is a SINE retrotransposon with basal and transcription factor-inducible insulator activity
B1-X35S has consensus-binding sites for AHR (XRE) and SLUG and SNAIL (E-box) (Supplemental Fig. 1A). Genomic analysis showed that B1-X35S has a prevalence of 14% in proximal promoters (<10 kb), 18% in distal promoters (10–50 kb), 48% in nonpromoter gene sequences, and 20% in any other genomic locations, additively giving a 32% occurrence in upstream gene regulatory regions (Supplemental Fig. 1B). To investigate the potential insulator activity of B1-X35S, we performed enhancer-blocking assays (EBA) in human HEK 293 cells. EBA revealed that B1-X35S has a potent intrinsic insulator activity, similar to that of the chicken 1.2-kb 5′HS4 beta-globin insulator (Fig. 1A; Recillas-Targa et al. 1999). Mutation of the XRE site (B1-Xmut35S) reduces insulation, while mutation of the E-box does not have a significant effect (Fig. 1A). Unlike AHR, basal levels of SLUG and SNAIL are very low in HEK 293 cells (Supplemental Fig. 1C), possibly accounting for the lack of effect of single E-box mutation under basal cell conditions. However, double XRE+E-box mutation (B1-Xmut35Smut) reproducibly reduced the insulation effect of single XRE mutation (Fig. 1A), suggesting functional interactions between factors bound at these sites.

B1-X35S is a SINE with insulator activity. (A) Wild-type B1-X35S or its mutant forms Xmut35S (AHR site mutated), X35Smut (E-box mutated), and Xmut35Smut (both sites mutated) were transiently transfected and their insulator activity analyzed by EBA. The constructs are illustrated at the top of the figure. Data are presented as fold-enhancer blocking activity normalized to the reference pELuc vector. (B) Occupancy of SLUG and SNAIL on B1-X35S was analyzed by qChIP after transient transfection of either protein. (C) Effects of AHR expression on SLUG and SNAIL recruitment to B1-X35S were determined by qChIP after transient transfection of AHR-specific shRNA or scrambled shRNA as negative control. (D) Simultaneous presence of AHR and SLUG on B1-X35S was analyzed by sequential qChIP. A first ChIP used anti-SLUG antibody and the immunoprecipitated DNA was re-ChIPed with anti-AHR antibody. Re-ChIP for GAPDH provides a negative control. Data were quantified with respect to input DNA from the first ChIP. Results for Dad1 and Tbc1d1 are shown. (E) The effect of AHR, SLUG, and SNAIL expression on the insulator activity of wild-type B1-X35S was analyzed by EBA and quantified as above. (F) The experiment in E was done using the double-mutant B1-Xmut35Smut. Human HEK 293 cells were used in A, E, and F, while mouse Hepa-1 cells were used in B, C, and D. (B,C) The average data from the five B1-X35S containing genes Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1. Individual results for each gene and experimental condition (B,C) are detailed in Supplemental Table 1. Data are shown as mean ±SD. (*) P = 0.033 and (**) P = 0.009 with respect to B1-X35S.
SLUG and SNAIL might compete to bind B1-X35S because they share the same response element. To test this, mouse Hepa-1 cells were transiently transfected with SLUG or SNAIL and chromatin immunoprecipitation (ChIP) was used to assay their binding to the promoter of five B1-X35S-containing genes (Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1; see Supplemental Fig. 1D for location). SLUG expression increased its own binding to B1-X35S and reduced that of SNAIL, while the converse was true for SNAIL expression (Fig. 1B). Consistent with the EBA results, reducing basal AHR levels with a specific shRNA severely impaired binding of both SLUG and SNAIL to B1-X35S in vivo (Fig. 1C), indicating that AHR facilitates SLUG and SNAIL recruitment to B1-X35S-containing promoters (individual results for each gene and experimental condition shown in Fig. 1B,C are detailed in Supplemental Table 1). Consistent with this, sequential ChIP (re-ChIP) demonstrated that AHR co-occupies with SLUG the B1-X35S element of Dad1 and Tbc1d1 (Fig. 1D).
As expected, individual increases in AHR or SLUG expression raise the insulator activity of B1-X35S, while coexpression of both has an additive effect, further strengthening insulation (Fig. 1E). Surprisingly, SNAIL expression had little effect on the EBA (Fig. 1E), suggesting that even though it can bind B1-X35S, it does not promote insulation. We therefore focused the remainder of the study on AHR and SLUG. The effects of these proteins require their recognition motifs, as transfection of AHR, SLUG, or SNAIL did not significantly alter the lower basal EBA of the double-mutant B1-Xmut35Smut transposon (Fig. 1F).
B1-X35S has insulator activity in zebrafish in vivo
Zebrafish has been established as a powerful model to analyze cis-regulatory genomic elements in vivo (Bessa et al. 2009). Zebrafish is a good model because it expresses three functional forms of AHR (AhR1a, the ortholog of mammalian AHR, AhR1b, and AhR2) (Mathew et al. 2009) and an ortholog of SLUG (Katoh 2005). B1-X35S and its mutant version B1-Xmut35Smut were cloned in the enhancer+promoter-driven GFP vector, as indicated in Figure 2A. These constructs were microinjected into one-cell zebrafish embryos and GFP expression quantified in CNS (enhancer-dependent and susceptible to insulator regulation) and somites (muscle promoter-driven and insulation-independent). B1-X35S has a potent insulator activity in zebrafish, down-regulating CNS enhancer-driven GFP expression (Fig. 2A). Mutation of AHR and SLUG-binding sites significantly reduced insulation (Fig. 2A). GFP signals in CNS and somites were quantified in each individual animal as the GFP ratio in muscle/CNS (Supplemental Fig. 2). The inter-individual variation is due to the integration of the transgene at different genomic locations in different somatic cells—they are not stable lines resulting from a unique integration in the germ line. The median fluorescence ratios revealed that wild-type B1-X35S, but not the double mutant, triggers a strong insulation response (Fig. 2B). Thus, B1-X35S is a B1 SINE with insulator activity in vivo.
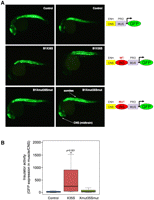
B1-X35S has insulator activity in zebrafish. (A) B1-X35S and mutant B1Xmut35Smut in the enhancer+promoter-driven GFP vector, as illustrated, were microinjected into one-cell zebrafish embryos. Each animal was photographed and GFP signals quantified in CNS and somites. Measurements of individual zebrafish are shown in Supplemental Figure 2. (B) Insulator activity was calculated as the median GFP signal in muscle/CNS for each experimental condition. Data are shown as mean ±SD.
AHR and SLUG modulate Pol III binding to B1-X35S
Pol III and Pol II transcription are implicated in insulation (Valenzuela and Kamakaka 2006; Lunyak et al. 2007; Lunyak 2008). Like canonical B1 repeats, B1-X35S has consensus A and B boxes for binding transcription factor TFIIIC, which then recruits transcription factor TFIIIB and Pol III (Supplemental Fig. 1A). DNA-binding affinity assays confirmed that Pol III and its assembly factor TFIIIC can bind B1-X35S in vitro (Fig. 3A). Mutation of the XRE element increases binding of both, while E-box mutation does not have a significant effect. Consistent with the EBA response (Fig. 1), XRE+E-box double mutation additively promotes Pol III and TFIIIC binding to B1-X35S (Fig. 3A). This can be explained if binding of AHR and SLUG interferes with binding of TFIIIC and, hence, Pol III. To further test this, Hepa-1 cells were transfected and occupancy was analyzed by qChIP of five B1-X35S loci. Increased expression of AHR, with or without SLUG, strongly suppresses binding to B1-X35S by Pol III, TFIIIC, and TFIIIB (Fig. 3B; Supplemental Table 1). These effects are specific, since histone H3 binding to B1-X35S is unaltered (Fig. 3C), as is Pol III binding to its unrelated target tRNALeu gene (Fig. 3D). AHR or SLUG expression does not alter Pol III protein levels in Hepa-1 cells (Supplemental Fig. 3).

Pol III binds B1-X35S: role of AHR and SLUG. (A) In vitro binding of Pol III and TFIIIC to B1-X35S, B1-Xmut35S, B1-X35Smut, or B1-Xmut35Smut was analyzed by DNA-binding affinity assays. (B) qChIP to address whether AHR and SLUG expression block Pol III recruitment to B1-X35S in vivo after transient transfection of Hepa-1 cells with AHR alone or with SLUG. (C,D) AHR was expressed with or without SLUG and their effects on histone H3 binding to B1-X35S (C) or Pol III binding to tRNALeu genes (D) determined by qChIP. (B,C) Average data from five B1-X35S-containing genes (Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1). Data are shown as mean ±SD.
B1-X35S is transcribed by Pol III, and AHR enhances transcription by promoting exchange of Pol III by Pol II
Although SLUG inhibits B1-X35S transcription in vitro, AHR produces an unexpected increase (Fig. 4A). In both cases, the responses depend on the respective binding motifs. SLUG ablates the effect of AHR on B1-X35S transcription, perhaps because AHR favors binding of inhibitory SLUG (see Figs. 1C, 3A,B). The positive effect of AHR on B1-X35S transcription was also found in vivo in transfected Hepa-1 (Fig. 4B). Therefore, AHR expression increases B1-X35S transcription despite inhibiting Pol III binding.

B1-X35S is transcribed by Pol III and AHR increases its transcription with exchange of Pol III by Pol II. (A) IVT using extracts of Hepa-1 cells transiently transfected with empty vector, AHR, or SLUG. Wild-type B1-X35S and mutants B1-Xmut35S and B1-X35Smut were used as templates. (B) B1-X35S expression analyzed by qRT–PCR using total RNA from Hepa-1 cells after transfection with empty vector or AHR. Concentrations of 20 μg/mL α-amanitin (α-AMA) or 5 μg/mL actinomycin-D were added where indicated. (C) The effect of AHR on Pol II binding to wild-type B1-X35S in Hepa-1 cells was analyzed by qChIP. (D) IVT of B1-X35S with extracts of Hepa-1 cells transfected with empty vector or AHR. A total of 10 μM tagetitoxin (TGT) or 3 μg/mL α-AMA were included where indicated. Percentages indicate the inhibition caused by each chemical relative to transcription in corresponding vehicle-treated reactions. (E) IVT as indicated in D using the mutant B1-Xmut35S. (F) IVT using wild-type B1-X35S and extracts of immortalized fibroblasts from AHR-null mice (T-FGM AhR−/−). (C) Average data from the five B1-X35S containing genes Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1. Data are shown as mean ±SD.
Some B2 SINEs carry active Pol II promoters (Ferrigno et al. 2001). Furthermore, Pol II-dependent transcription may influence B2 insulator function (Lunyak et al. 2007). We therefore tested whether AHR activates B1-X35S transcription through Pol II. qChIP revealed that AHR expression increases Pol II binding to B1-X35S (Fig. 4C; Supplemental Table 1) without raising Pol II protein levels (Supplemental Fig. 4).
In Hepa-1 cells, the Pol III inhibitor tagetitoxin (TGT) reduces B1-X35S transcription by 70%, whereas α-amanitin (α-AMA) at a concentration specifically inhibiting Pol II (Rollins et al. 2007) decreases it by 50% (Fig. 4D). An increase in AHR expression attenuates the inhibitory effect of TGT to 45%, but increases the inhibition caused by α-AMA to 80% (Fig. 4D). These data indicate that both Pol III and Pol II participate in B1-X35S transcription, with Pol III contributing more than Pol II. However, an increase in AHR expression changes the ratio in favor of Pol II (Fig. 4D). Indeed, α-AMA efficiently inhibits in vivo the increase in B1-X35S transcription caused by AHR expression (Fig. 4B), further supporting that such effect is Pol II dependent. Comparison of the inhibition caused by α-AMA and the general transcriptional blocker actinomycin D also indicates that Pol II has a significant contribution to AHR-dependent B1-X35S transcription (Fig. 4B). The importance of AHR in this Pol III–Pol II exchange is supported by two additional sets of experiments. First, XRE mutation (B1-Xmut35S) abolishes the inhibitory effect of α-AMA, but not that of TGT (Fig. 4E). Second, B1-X35S transcription in extracts of cells lacking AHR expression (T-FGM AhR−/−) is insensitive to α-AMA but remains susceptible to TGT (Fig. 4F). It seems that the basal insulator activity of B1-X35S is maintained by Pol III transcription primarily, while the enhanced insulation caused by AHR relies, at least in part, on an increase in transcription due to a switch to Pol II from Pol III.
Pol III and Pol II transcribe B1-X35S in the same direction
Some B2 SINEs carry a Pol II promoter that is orientated in the opposite direction to the Pol III promoter (Ferrigno et al. 2001). Similarly, the insulator activity of a B2 SINE involves its transcription from opposite strands by Pol III and Pol II (Lunyak et al. 2007). To clarify the direction of transcription of B1-X35S, we used strand-specific RT–qPCR with sense and antisense primers, as schematized in Figure 5A. The level of RT-generated products was clearly detected in the antisense, but not the sense direction, suggesting that most expression is from one strand. Pol II inhibition with α-AMA decreased the amount of antisense RT product by 57%, but had no effect on the low levels of sense RT product (Fig. 5A). Our data therefore suggest that Pol III and Pol II transcribe B1-X35S in the same direction (Fig. 5A, solid arrows).

The same strand of B1-X35S is transcribed by Pol III and Pol II. (A) IVT of B1-X35S analyzed using direction-specific sense and antisense with extracts of Hepa-1 cells treated with or without 3 μg/mL α-AMA. A negative qPCR control was performed in the absence of RT template (−RT). (B) Binding of Pol III and Pol II on B1-X35S was determined by re-ChIP. First ChIP used anti-Pol III antibody, and the resulting DNA was immunoprecipitated again with anti-Pol II or anti-TFIIIC (positive control) antibodies. Re-ChIP for GAPDH was used as negative control. Data are presented as percentage of input from the first ChIP. (C) Pol III binding in vitro to B1-X35S A+B mutant was determined by DNA affinity assays with anti-Pol III RPC32 antibody. (D) EBA performed as for Figure 1A using wild-type B1-X35S and B1-X35S A+B mutant with or without AHR transfection. (B) Average data from five B1-X35S containing genes (Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1). Data are shown as mean ±SD.
Occupancy of B1-X35S by Pol III and Pol II was further analyzed by sequential ChIP assays (Re-ChIP). Immunoprecipitation of Pol II on B1-X35S DNA from Pol III ChIPs revealed that Pol III prevails under basal conditions over Pol II (Fig. 5B). Mutation of the A and B boxes impairs Pol III binding to B1-X35S in DNA-binding affinity assays (Fig. 5C). EBA experiments showed that A+B mutation severely impairs the intrinsic insulator activity of B1-X35S (Fig. 5D). This supports an important role for Pol III in basal SINE-mediated insulation. Moreover, mutation of the A and B boxes abolished the increase in insulator activity induced by AHR overexpression, suggesting that the enhanced insulation associated with Pol II requires the Pol III promoter. Overall, these data strongly suggest that high AHR levels promote the release of Pol III and the engagement of Pol II on B1-X35S, with a concomitant increase in transcription and in insulator activity. B1-X35S does not have a canonical TATA box, but it contains a similar sequence (5′-TTAAT-3′). Whether this is the binding site for Pol II remains to be explored.
B1-X35S insulation correlates with epigenetic marks of heterochromatin and with CTCF binding, and both properties are modulated by AHR
To analyze the relationship between B1-X35S and epigenetic marks that might sustain its insulator function, we analyzed by qChIP the profile of heterochromatin marks in histone H3 within a −1000 to +1500 region upstream and downstream from B1-X35S in Dad1, Lpp, and Cabin1 (Fig. 6A; Supplemental Table 2). Histone H3 trimethyl lysine 9 (H3K9me3) (Fig. 6B) and trimethyl lysine 27 (H3K27me3) (Fig. 6C) marks are lower upstream of B1-X35S and increase downstream from the transposon. AHR+SLUG expression enhances significantly H3K9me3 and H3K27me3 levels downstream from the repeat (Fig. 6B,C).

B1-X35S establishes a heterochromatic epigenetic mark that responds to AHR. The region from 1000 bp upstream (−1000) to 1500 bp downstream (+1500) from B1-X35S was analyzed for chromatin marks by qChIP in Hepa-1 cells with or without transfection of AHR+SLUG. (A) Location of B1-X35S in the promoter region of Dad1, Lpp, and Cabin1 and of the PCR fragments analyzed is indicated. (B) H3K9me3 status. (C) H3K27me3 status. Data were normalized to the amount of H3 immunoprecipitated. (D) In vivo binding of CTCF and PARP1 to B1-X35S was analyzed by qChIP in Hepa-1 cells with or without transfection of AHR+SLUG. PARP1 inhibitor 3-amino benzamide (3-ABA, 5 mM) was added for 24 h where indicated. Average results for genes Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1 are shown. Experiments were performed in Hepa-1 cells and data are shown as mean ±SD.
CTCF is a transcription factor with a role in the control of genomic boundaries and insulators (Bushey et al. 2008). Binding of CTCF to insulator sequences correlates with an increase in heterochromatin marks such as H3K27me3 (Han et al. 2008; Li et al. 2008). We therefore asked whether CTCF binds B1-X35S in vivo. qChIP showed that CTCF binds B1-X35S and that such binding is significantly enhanced by AHR expression, but only marginally by SLUG expression (Fig. 6D). Consistently, with the accumulation of H3K27me3, coexpression of AHR+SLUG additively increases CTCF binding to B1-X35S. CTCF is activated by poly-ADP-ribose polymerase (PARP1)-dependent parylation (Witcher and Emerson 2009) and, accordingly, PARP1 binding to B1-X35S follows a pattern close to that of CTCF (Fig. 6D). The PARP1 inhibitor 3-amino benzamide (3-ABA) (Witcher and Emerson 2009) significantly reduces CTCF binding to B1-X35S in vivo, suggesting that parylation facilitates the interaction (Fig. 6D). On the contrary, histone H3, a nucleosomal protein not expected to interact with CTCF nor PARP1, binds B1-X35S in an AHR- and SLUG-independent manner (Fig. 3C). Therefore, the insulator activity of B1-X35S may involve an increase in heterochromatin content downstream of the transposon, perhaps through recruitment of parylated CTCF.
It has been suggested that insulators and SINEs have a role in chromatin compartmentalization. Based on the accumulation of heterochromatic marks downstream of B1-X35S, we performed a comparative epigenomic analysis (from NCBI GEO repository GSE12241 (Mikkelsen et al. 2007) for H3K9me3 in mouse embryonic stem (ES) cells vs. embryonic fibroblasts (MEF). We focused on a −500 to +500 region flanking B1-X35S in 75 instances. H3K9me3 is markedly enriched downstream of B1-X35S in ES cells, but not in MEFs (Fig. 7A), suggesting a cell type-dependent function in setting heterochromatin domains. To address this further, the variance in H3K9me3 content was calculated as the Manhattan distance in a 500-bp region downstream of B1-X35S using 700 common instances for ES and MEF (Fig. 7B). It was found that the ES/MEF distance for H3K9me3 markedly increased downstream of B1-X35S (B1 to +500). These results agree with our experimental data (see Figs. 6B,C, 7A) and suggest a role for B1-X35S in establishing heterochromatic regions that vary with cell type, perhaps depending on the differentiation status. In contrast, heterochromatic H3K9me3 regions were not differentially established by murine ID SINE elements (Fig. 7B) in ES vs. MEF, indicating that SINE elements differ in their ability to modulate chromatin compaction and probably to insulate gene expression. Indeed, EBA assays reveal that insulation for an ID element is significantly lower than that to B1-X35S (Supplemental Fig. 5).
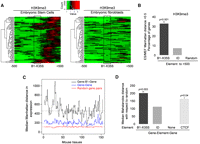
Distinct patterns of heterochromatin and gene expression upstream and downstream from B1-X35S. (A) H3K9me3 analyzed in mouse ES cells and MEFs at 75 B1-X35S loci between −500 bp (upstream) and +500 bp (downstream) from the SINE (box). ChIP-seq data were taken from NCBI GEO repository (GSE12241) (Mikkelsen et al. 2007). Red indicates higher and green lower H3K9me3 levels. (B) H3K9me3 levels downstream of B1-X35S and SINE ID elements (element to +500) in 700 instances common to ES and MEF cells were analyzed. Data are presented as the ES/MEF Manhattan distance ratio. A larger distance indicates higher H3K9me3 content in ES cells. Random sequences were also analyzed as negative controls. (C) The effect of B1-X35S on the differential expression of flanking genes was analyzed in the following groups (700 gene pairs for each): gene pairs having a B1-X35S between them (gene-B1-gene); gene pairs lacking B1-X35S (gene-gene); randomly located gene pairs (random). The difference in expression for each pair in every group was analyzed in 150 tissues of the mouse gene expression atlas and the values obtained represented as the median Manhattan distance in expression. Higher values represent larger differences between adjacent genes. The mean distance for each condition is indicated by a horizontal dashed line. (D) The gene–B1X35S-gene, gene–ID-gene, and gene–gene groups were analyzed using the Mahalanobis algorithm. The impact of the binding sequence for CTCF on the expression of contiguous genes (gene–CTCF-gene) was also determined using ChIP-seq data from the NCBI GEO repository (GSE11431) (Chen et al. 2008).
We sought to determine whether the presence of B1-X35S between adjacent genes induces divergent patterns of gene expression. We configured three gene groups (700 gene pairs for each condition) in which: (1) two adjacent genes have a B1-X35S in between; (2) two genes are adjacent without a B1-X35S; or (3) the two genes are randomly located. The median difference in expression for the two genes in each of the 700 pairs for each condition (e.g., gene–B1-gene, or gene–gene, or random gene pair) was obtained for 150 tissues of the mouse gene expression atlas (Fig. 7C). Manhattan distance analysis revealed that the presence of B1-X35S between contiguous genes significantly increases their median difference in expression (black), as compared with adjacent genes lacking any SINE (blue) or to randomly located gene pairs (red). The use of the Mahalanobis algorithm gave a similar result, indicating that B1-X35S accentuates the difference in expression between contiguous genes (Fig. 7D). While the presence between adjacent genes of the binding site for CTCF also increases their difference in expression, the insertion of ID SINEs does not have a significant effect (Fig. 7D). Thus, these data suggest that two contiguous genes separated by a B1-X35S element acquire different levels of expression, probably sustained by heterochromatin marks such as H3K9me3 at one side of the SINE.
We postulated that B1-X35S SINEs could have been selected and fixed at certain genomic locations due to their intrinsic capacities to attract AHR and SLUG and to recruit epigenetic modifications. To address this, we split the 14,000 known locations of B1-X35S into three categories, depending on whether this SINE is located at promoter regions (up to −10 kb from the transcription start site), further upstream (between −10 kb and −50 kb), or internally within the locus. These three gene sets (available upon request) were subjected to gene expression analysis with the Explain program (BioBase; http://biobase-international.com/pages/index.php?id=286) to extract transcriptional networks that share a common subset of genes. Signaling networks were identified for the regulators of survival, proliferation, and apoptosis AKT and p62DOK1 (Supplemental Fig. 6) for the modulators of the stress response, apoptosis, and angiogenesis SGK-1 and JNK-1 (Supplemental Fig. 7) and for the essential regulator of mitochondrial apoptosis caspase 6 (Supplemental Fig. 8).
The functional relevance of B1-X35S in the control of gene expression was also estimated by gene-ontology analysis. We found that the occurrence of B1-X35S at different genomic locations varies according to functional categories and is markedly under-represented in the promoters of genes involved in neurological disorders, sensory perception, and G protein-coupled receptors. Conversely, it is over-represented in the promoters of genes associated with ubiquitin ligase activity and ATP binding (Supplemental Table 3).
Discussion
A major finding of this work is that the recently identified SINE B1-X35S retrotransposon has potent intrinsic insulator activity in vitro and in vivo, and that this function can be enhanced by its interaction with the transcription factors AHR and SLUG. The involvement of these proteins in controlling insulation was unexpected and could have considerable biological significance. AHR is a key regulator of xenobiotic-induced carcinogenesis (Shimizu et al. 2000; Marlowe and Puga 2005) and has a role in cell proliferation (Barouki et al. 2007), immune T cell differentiation (Quintana et al. 2008), migration (Diry et al. 2006; Carvajal-Gonzalez et al. 2009), and angiogenesis (Roman et al. 2009). SLUG and SNAIL are important in promoting migration during development and disease by inducing EMT, the epithelial-to-mesenchymal transition (Thiery et al. 2009). Although SLUG and SNAIL are closely related and recognize the same consensus sequence, SNAIL did not stimulate insulation despite its efficient binding to B1-X35S, suggesting differences in the regulatory interactions that modulate insulation. AHR and SLUG have an additive effect on B1-X35S insulation, which might be relevant to pathological conditions such as metastatic cancer, given the migration-promoting activity of AHR and SLUG in mesenchymal cells. Consistent with the insulator activity, AHR and SLUG cooperate to repress transcription of B1-X35S-containing genes (Roman et al. 2008), possibly because AHR promotes SLUG binding to B1-X35S.
As for the B2 SINE at the growth hormone locus (Lunyak et al. 2007), B1-X35S-dependent insulation is associated with transcription across the transposon. However, two main differences were found with respect to the B2 case: First, Pol III and Pol II transcribe B1-X35S from the same strand in the downstream direction and, second, AHR increases B1-X35S transcription (which is otherwise mainly dependent on Pol III) by an exchange mechanism that recruits Pol II and releases Pol III from the SINE. The AHR-dependent increase in B1-X35S transcription by Pol II is associated with enhanced insulator activity, indicating that SINE transcription correlates with the degree of insulation. Recruitment of Pol II to B1-X35S requires the A and B box Pol III promoter elements and mutation of these abolishes the inducing effect of AHR on B1-X35S insulation. Pol III and its associated factors bound to the A and B boxes may establish epigenetic marks that allow subsequent recruitment of Pol II (Kenneth et al. 2007; Barski et al. 2010; Moqtaderi et al. 2010; Raha et al. 2010). It is striking that AHR induces the replacement of Pol III by Pol II at B1-X35S. We are not aware of a precedent for this behavior.
It seems paradoxical that replacement of Pol III by Pol II increases transcription of B1-X35S. Pol III transcription is generally very efficient due to a high rate of re-initiation, in which the polymerase is recycled to the start site without dissociating from the template DNA (Dieci and Sentenac 2003). For example, tRNA and 7SL RNA genes utilize this mechanism to maintain rapid transcription (Ferrari et al. 2004). Indeed, the four 7SL gene copies in humans are sufficient to maintain transcript levels of ∼106 copies per cell (Liu et al. 1994). However, we found that Pol III occupancy is much lower on B1 SINEs than on Pol III templates that are actively transcribed, such as 7SL or tRNA genes (Supplemental Fig. 9). This reflects a defect subsequent to TFIIIC binding, as the ratio of Pol III/TFIIIC ChIP signals is significantly lower on B1 when compared with 7SL (P = 0.002 by Student t-test). Therefore, our data suggest that Pol III recruitment is relatively inefficient at B1 SINEs, which might explain why replacement of Pol III with a Pol II-dependent system can result in increased expression. Replacement may occur by AHR interfering with TFIIIC binding to the A and B boxes.
Transposable elements and insulators are closely linked to the epigenetic regulation of the genome (Slotkin and Martienssen 2007; Bushey et al. 2008; Tomilin 2008). B1-X35S shares with other insulators, such as the Su(Hw), Mod(mdg) (Gerasimova and Corces 1998), and the growth hormone B2 SINE (Lunyak et al. 2007), the characteristic of heterochromatin marks capable of regulating cis-acting genes. An elevation of heterochromatin-associated marks was found downstream of B1-X35S, and this pattern was increased by AHR+SLUG binding, suggesting that these transcription factors determine the recruitment of histone methyltransferases to specific regions of the genome adjacent to the transposon. This is consistent with previous studies showing that nucleosomes at transposons are often enriched in heterochromatin marks like H3K9me3 (Martens et al. 2005), and that a reporter gene introduced in the genome of ES cells by LINE-1 retrotransposons is rapidly repressed by chromatin modifications at the insertion site, while ES cell differentiation attenuates the silencing induced by LINE-1 (Garcia-Perez et al. 2010). The ability of B1-X35S to provide a potential heterochromatin barrier could have substantial functional impact across the genome. Because it displays markedly different H3K9me3 profiles in ES cells and fibroblasts, the insulator activity of B1-X35S may regulate gene expression during development. Location of a B1-X35S SINE between two adjacent genes correlates with their differential expression in most adult mouse tissues, supporting the involvement of B1-X35S in establishing distinct expression domains for flanking genes, possibly by modulating chromatin. DNA methylation of B1-X35S could also play a role in insulation, since changes in methylation of CpG dinucleotides flanking human Alu repeats are implicated in ependymomas (Xie et al. 2010). In addition, we have shown that the insulator activity of B1-X35S involves binding of parylated CTCF, an established insulator-binding protein involved in loop formation and the recruitment of repressive complexes to chromatin (Han et al. 2008; Li et al. 2008). AHR-mediated binding of CTCF/PARP1 to B1-X35S-containing genes could contribute to their transcriptional repression (Roman et al. 2008). A similar mechanism has been proposed for the transcriptional regulation of p16ink4a expression in human tumor cells (Witcher and Emerson 2009).
The clustering of genes containing B1-X35S elements revealed the existence of transcriptional networks controlling the cell cycle, proliferation, apoptosis, and cancer. Both AHR and SLUG have been implicated in cell growth and proliferation, apoptosis, and migration, thus supporting their functional interaction with the AKT-1, p62DOK1, SGK-1, JNK-1, and caspase 6 pathways. Indeed, AHR seems to regulate dioxin-induced migration through the JNK pathway in human breast tumor cells (Diry et al. 2006), while AHR deficiency results in lower AKT activation in mouse fibroblasts (Mulero-Navarro et al. 2005). Given the interaction between these transcriptional networks and AHR and SLUG, we envisage that most B1-X35S-linked genes may normally be repressed by these factors, perhaps because increased SINE transcription by Pol II enhances the insulator and chromatin barrier activities. In the absence of AHR and SLUG, B1-X35S transcription by Pol II would be reduced and its insulator activity diminished, eventually resulting in gene derepression and in altered cellular homeostasis. B1-X35S seems to be restricted to the mouse genus (Roman et al. 2008) and caution should be taken in extrapolating the insulator activity of AHR and SLUG to other species, since the profile of occupancy of binding sites for transcription factors (e.g., OCT-8 and NANOG but not CTCF) in transposable elements vary widely between mouse and human ES cells (Kunarso et al. 2010). Interestingly, B1-X35S has insulator activity in both fish and human cells. While maintaining this property in different experimental systems helps establish the role of B1-X35S as a genome-wide insulator, our conclusions are not necessarily applicable to repetitive elements and expression patterns of AHR and SLUG in other species. Certainly, a different repetitive element such as the murine SINE ID has significantly lower insulator activity than B1-X35S.
In summary, we propose a model for the insulator activity of B1-X35S SINE retrotransposons involving AHR and SLUG, Pol III, and Pol II, and the epigenetic regulation of chromatin (Fig. 8). Under basal conditions, Pol III is predominant at B1-X35S and provides its intrinsic insulator activity. Elevated AHR expression triggers recruitment of Pol II and the displacement of Pol III from the SINE, raising B1-X35S transcription and enhancing insulation. AHR increases heterochromatin marks downstream from B1-X35S, which establish a chromatin barrier that contributes to the inhibition of neighboring genes. The insulator activity of B1-X35S also seems to involve parylated CTCF. Although further studies will be required to validate this model at specific genomic loci, it has potential relevance genome wide. It is now apparent that AHR and SLUG have unexpected functions beyond their classical role in transcription. These molecular events may have an impact on development and cell physiology in health and disease.
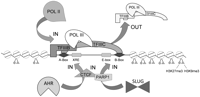
Proposed model for the regulation of the B1-X35S insulator. Arrows indicate entry and exit of proteins. Small gray circles indicate H3K27me3 and small gray triangles H3K9me3. Binding sites for AHR, SLUG, SNAIL, and TFIIIC are indicated. Potential involvement of parylated CTCF is also indicated.
Methods
Reagents, antibodies, plasmids, and cell lines
3-amino benzamide, α-amanitin, and anti-actin antibody were purchased from Sigma. Tagetitoxin was from Epicentre Biotechnologies. Protein A/G Plus Agarose, anti-SLUG (sc-10437), anti-Pol II (N-20), and anti-Pol III (RPC32) antibodies were from Santa Cruz Biotechnology. Antibodies to histone H3 (H3), H3K9me3, H3K27me3, and H3K4me3 were purchased from Diagenode. Other commercial antibodies used include anti-AHR MA1-514 (Affinity Bioreagents), anti-GAPDH (Cell Signaling), and anti-PARP1 (Roche). Polyclonal antisera against Pol III (RPC155), TFIIIB (Brf1), and TFIIIC (102kD and 110kD) have been described (Kenneth et al. 2008). Anti-SNAIL and anti-CTCF antibodies were kindly provided by Dr. Antonio Garcia de Herreros and Dr. Felix Recillas-Targa, respectively.
B1-X35S-containing plasmids were made by sequential PCR. A first amplification reaction was performed using long overlapping primers (B1-X35 and B1-35S and their Xmut, Smut, and ABmutants indicated in Supplemental Table 5 and Supplemental Fig. 1A) covering the full-length B1-X35S retrotransposon. The products obtained were then used as templates in a second PCR reaction performed using a pair of common flanking primers (Flank-F and Flank-R in Supplemental Table 5). The resulting products were A/T cloned into pGEM-T for further use (Promega).
Mouse Hepa-1 cells (Interlab Cell Line Collection ATL98016) were cultured in α-MEM (Invitrogen) as indicated (Roman et al. 2008). Immortalized fibroblasts from AhR+/+ and AhR−/− mice were used as previously (Carvajal-Gonzalez et al. 2009).
Transfection and Western blotting
Hepa-1 cells were transiently transfected in a MicroPorator (DigitalBio) using a single pulse of 1400 V for 30 msec, following the manufacturer's recommendations. Protein expression and the analysis of protein–DNA interactions in vitro were by Western blotting as indicated (Roman et al. 2008).
Quantitative Chromatin Immunoprecipitation (qChIP)
qChIP was performed as described (Roman et al. 2008; Carvajal-Gonzalez et al. 2009). Positive controls were performed using input DNAs, whereas negative controls included binding reactions in the absence of specific antibodies. To quantify changes in protein binding to DNA, all amplifications were performed by real-time PCR (qPCR) using IQ-SYBR Green in an iCycler equipment (Roman et al. 2009). Data are presented as percentage of DNA input in the antibody-containing immunoprecipitates minus the percentage of DNA input in the corresponding negative controls. Bars represent the average amplification of B1-X35S elements located in the upstream promoter region of the genes Dad1, Lpp, Cabin1, Tbc1d1, and Rtl1. Individual data for each gene and experimental condition are provided in Supplemental Table 1. In experiments mapping histone H3 modifications upstream and downstream of B1-X35S only Dad1, Lpp, and Cabin1 were analyzed. Individual data for each gene and experimental condition are provided in Supplemental Table 2.
Sequential ChIP (re-ChIP)
Sequential ChIP was used to analyze simultaneous binding of two proteins to a common DNA region. Re-ChIP was by q-PCR following a published protocol (Metivier et al. 2008) using primers for B1-X35S elements located in promoters of the genes listed in Supplemental Table 4. GAPDH was immunoprecipitated as negative control for the second ChIP. Results are presented as percentage of DNA in the primary immunoprecipitates.
Enhancer blocking assay (EBA)
Enhancer blocking assay to address the insulator activity of wild-type and mutant B1-X35S used the pELuc plasmid previously described (Lunyak et al. 2007). B1-X35S elements were cloned between the CMV enhancer and the promoter (XhoI) or upstream of the CMV enhancer (PstI). EBA was performed by transfecting the constructs into human embryonic kidney HEK 293 cells as reported (Lunyak et al. 2007). Data are presented as fold-enhancer blocking activity normalized to the value achieved by the basal pELuc vector. The 1.2-kb chicken 5′HS4 beta-globin insulator element was used as positive control. Similarly, the internal II/III boxes from the chicken 5′HS4 beta-globin insulator element, both wild type and mutated, were used as positive and negative controls, respectively (Recillas-Targa et al. 1999). See schemes in Figure 1.
In vivo insulator activity in zebrafish
The insulator activity of wild-type and mutant B1-X35S was also analyzed in vivo by microinjection of the constructs drawn in Figure 2 into one-cell zebrafish embryos as described (Bessa et al. 2009). Briefly, B1-X35S elements were cloned in a GFP reporter that allows quantification of enhancer-blocking activity by measuring the ratio of fluorescence in somites (driven by a cardiac actin promoter) vs. fluorescence in the central nervous system (regulated by the Z48 midbrain neuronal enhancer). Individual specimens were analyzed 36 h after microinjection. At least 20 zebrafish were photographed and quantified for each condition in two independent experiments. LaserPix (Bio-Rad) image analysis software was used for quantification.
Epigenomic bioinformatic analysis
Genomic data for H3K9me3 in embryonic stem cells (ES) and mouse embryonic fibroblasts (MEF) were retrieved from NCBI GEO repository (GSE12241) (Mikkelsen et al. 2007). Genomic location of all B1-X35S elements was obtained using a modified version of our previously described script (Roman et al. 2008). We selected B1-X35S elements having significant differences in H3K9me3 in a −500 bp to +500 bp window from the transposon position (paired t-test <0.001 between ES and MEF). Hierarchical clustering (complete linkage) of ES cells and heat maps of ES and MEF cells were done using R statistical package. The effect of B1-X35S on the expression of adjacent genes was analyzed in 150 mouse tissues from the mouse gene expression atlas (Su et al. 2002). The Manhattan distance algorithm was used to quantify differences in H3K9me3 content upstream and downstream of B1-X35S in ES and MEF cells and to determine changes in the expression of pairs of genes located at both sides of the transposon. In some cases, the Mahalanobis distance was also calculated to address changes in gene expression due to the presence of either B1-X35S or CTCF. Genomic data for the presence of CTCF binding sites was retrieved from the NCBI GEO repository (GSE11431) (Chen et al. 2008).
DNA-binding affinity assays
Protein binding to B1-X35S in vitro was analyzed by DNA-binding affinity as described (Roman et al. 2008). The primers indicated in Supplemental Table 5 were biotin labeled and used to amplify by PCR the wild-type and mutant forms of B1-X35S as above. Binding reactions used streptavidin μMACS Factor Finder kit (Miltenyi Biotec) following the manufacturer's instructions. Proteins were detected by Western blotting.
In vitro transcription (IVT)
IVT was performed as previously described (Francis et al. 2005) with some modifications. Wild-type or mutant pGEM-T-B1-X35S constructs were used as templates. Hepa-1 protein extracts were pre-mixed with vehicle or with specific inhibitors of Pol II (3 μg/mL α-amanitin, α-AMA) (Rollins et al. 2007) or Pol III (10 μM tagetitoxin, TGT) (Perl et al. 2000). Treated extracts were then mixed with templates and IVT started by addition of 0.4 mM rNTPs. Reactions proceeded for 60 min at 30°C. After treatment with DNase I for 30 min, reactions were stopped and precipitated with ethanol. Purified RNA was subjected to RT–qPCR as described (Roman et al. 2009) using the primers in Supplemental Table 5. For strand-specific IVT, reverse transcription used sense (B1-Flank-F) or antisense (B1-X35S-R)-specific primers. Negative controls included reactions in the absence of protein, DNA, or RT product.
Statistical analyses
Experiments were performed three times with at least two replicates. Data are shown as mean ±SD. Statistical tests (unpaired and paired t-tests) were applied using the R statistical package. Wilcoxon test was applied for median-based analyses.
Acknowledgments
This work was supported by grants to P.M.F-S. from the Spanish Ministry of Science and Innovation (MICINN) (SAF2008-00462), the Junta de Extremadura (GRU09001), and the Red Temática de Investigación Cooperativa en Cáncer (RTICC) [RD06/0020/1016, Fondo de Investigaciones Sanitarias (FIS), Carlos III Institute, Spanish Ministry of Health]; by Grants to L.M. from the Spanish Ministry of Education and Science (BFU2006-12185) and MICINN (BIO2009-1297); and by Grants to J.L.G.-S. from the Spanish and Andalusian Governments (BFU2007-60042/BMC, Petri PET2007_0158, Proyecto de Excelencia CVI-3488 and CSD2007-00008). A.C.R. and F.J.G.-R. were supported by the MICINN, and E.M. was supported by CIBERER (ISCIII). All Spanish funding is co-sponsored by the European Union FEDER program. J.V.-A. and R.J.W. were funded by the Wellcome Trust and Cancer Research UK. The generous gifts of Dr. Antonio Garcia de Herreros (Institut Municipal d'Investigació Mèdica, Barcelona, Spain) (anti-SNAIL antibody and pcDNA-SNAIL), Dr. Felix Recillas-Targa (Instituto de Fisiología Celular, UNAM, Mexico) (anti-CTCF antibody), and Dr. Amparo Cano (Instituto de Investigaciones Biomédicas, Madrid, Spain) (pcDNA-SLUG) are greatly appreciated. The helpful comments and suggestions of Drs. R. Kamakaka, R. Ohlsson, and J.M. Merino are acknowledged.
Footnotes
-
↵8 Corresponding author.
E-mail pmfersal{at}unex.es; fax 34-924-289419.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.111203.110.
- Received June 2, 2010.
- Accepted December 16, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








