
Analysis of copy number variations among diverse cattle breeds
- George E. Liu1,13,
- Yali Hou1,2,
- Bin Zhu3,
- Maria Francesca Cardone4,
- Lu Jiang3,
- Angelo Cellamare4,
- Apratim Mitra2,
- Leeson J. Alexander5,
- Luiz L. Coutinho6,
- Maria Elena Dell'Aquila7,
- Lou C. Gasbarre1,
- Gianni Lacalandra7,
- Robert W. Li1,
- Lakshmi K. Matukumalli1,8,
- Dan Nonneman9,
- Luciana C. de A. Regitano10,
- Tim P.L. Smith9,
- Jiuzhou Song2,
- Tad S. Sonstegard1,
- Curt P. Van Tassell1,
- Mario Ventura4,
- Evan E. Eichler11,12,
- Tara G. McDaneld9 and
- John W. Keele9,13
- 1 USDA-ARS, ANRI, Bovine Functional Genomics Laboratory, Beltsville, Maryland 20705, USA;
- 2 Department of Animal and Avian Sciences, University of Maryland, College Park, Maryland 20742, USA;
- 3 Department of Bioengineering, University of Maryland, College Park, Maryland 20742, USA;
- 4 Department of Genetics and Microbiology, University of Bari, Bari 70126, Italy;
- 5 USDA-ARS, LARRL, Fort Keogh Miles City, Montana 59301, USA;
- 6 Departamento de Zootecnia, ESALQ-USP, Piracicaba SP 13418-900, Brazil;
- 7 Department of Animal Production, Faculty of Biotechnological Sciences, S. Prov. Casamassima, km 3-70010 Valenzano (Bari), Italy;
- 8 Bioinformatics and Computational Biology, George Mason University, Manassas, Virginia 20110, USA;
- 9 USDA-ARS, US Meat Animal Research Center, Clay Center, Nebraska 68933, USA;
- 10 Embrapa Pecuaria Sudeste, Sao Carlos–Sao Paulo, Rodovia Washington Luiz, km 234, Caixa Postal 339, CEP 13560-970, Brazil;
- 11 Deparment of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA;
- 12 Howard Hughes Medical Institute, Seattle, Washington 98195, USA
Abstract
Genomic structural variation is an important and abundant source of genetic and phenotypic variation. Here, we describe the first systematic and genome-wide analysis of copy number variations (CNVs) in modern domesticated cattle using array comparative genomic hybridization (array CGH), quantitative PCR (qPCR), and fluorescent in situ hybridization (FISH). The array CGH panel included 90 animals from 11 Bos taurus, three Bos indicus, and three composite breeds for beef, dairy, or dual purpose. We identified over 200 candidate CNV regions (CNVRs) in total and 177 within known chromosomes, which harbor or are adjacent to gains or losses. These 177 high-confidence CNVRs cover 28.1 megabases or ∼1.07% of the genome. Over 50% of the CNVRs (89/177) were found in multiple animals or breeds and analysis revealed breed-specific frequency differences and reflected aspects of the known ancestry of these cattle breeds. Selected CNVs were further validated by independent methods using qPCR and FISH. Approximately 67% of the CNVRs (119/177) completely or partially span cattle genes and 61% of the CNVRs (108/177) directly overlap with segmental duplications. The CNVRs span about 400 annotated cattle genes that are significantly enriched for specific biological functions, such as immunity, lactation, reproduction, and rumination. Multiple gene families, including ULBP, have gone through ruminant lineage-specific gene amplification. We detected and confirmed marked differences in their CNV frequencies across diverse breeds, indicating that some cattle CNVs are likely to arise independently in breeds and contribute to breed differences. Our results provide a valuable resource beyond microsatellites and single nucleotide polymorphisms to explore the full dimension of genetic variability for future cattle genomic research.
Over the last few years, bovine genomics has progressed rapidly generating a number of valuable resources, including a composite physical map (Snelling et al. 2007) and two independent genome assemblies (Btau_4.0 and UMD3; The Bovine Genome Sequencing and Analysis Consortium 2009; Zimin et al. 2009). These resources provide preliminary evidence for ruminant-specific variations in genes associated with lactation and immune responsiveness. The cattle research community has migrated from microsatellites to single nucleotide polymorphisms (SNPs) as the main measure of genetic variation in cattle, producing the first version of a cattle SNP map (The Bovine HapMap Consortium 2009) and the BovineSNP50 (>50,000 SNP probes) genotyping array (Van Tassell et al. 2008; Matukumalli et al. 2009). Their initial results indicate that during a rapid and recent decrease in effective population size from a very large ancestral population, detectable signatures of selection exist within the cattle genome due to domestication, selection, and breed formation. To accelerate livestock genetic improvement for milk and meat production, most ongoing efforts are focusing on whole-genome animal selection based on SNPs.
However, substantial progress has been made in understanding other forms of genetic variation, such as genomic structural variation, in other organisms including human (Iafrate et al. 2004; Sebat et al. 2004; McCarroll et al. 2006; Redon et al. 2006; Wong et al. 2007; Conrad et al. 2009), chimpanzee (Perry et al. 2006, 2008), rhesus monkey (Lee et al. 2008), mouse (Li et al. 2004; Adams et al. 2005; Snijders et al. 2005; Cutler et al. 2007; Graubert et al. 2007; She et al. 2008; Watkins-Chow and Pavan 2008), rat (Guryev et al. 2008), dog (Chen et al. 2009; Nicholas et al. 2009), and fruit fly (Emerson et al. 2008). Changes in DNA content and structure are a significant source of genetic and phenotypic variation among individuals (Feuk et al. 2006; Beckmann et al. 2007; Conrad and Antonarakis 2007; McCarroll and Altshuler 2007). These types of structural variations ranging from 1 kilobase (kb) to 5 megabase (Mb) comprised mainly of copy number variation (CNV in the form of large-scale insertions and deletions), as well as inversions and translocations. In humans, the Database of Genomic Variants (as of January 2010, http://projects.tcag.ca/variation/) contains ∼29,000 CNVs that correspond to over 8400 CNV regions identified in normal individuals. These data sets alone correspond to over 910 Mb of structurally variant DNA. More than 9000 genes have been mapped within or near regions of human structural variation. While SNPs are more frequent, CNVs involve more genomic sequences and have potentially more effects, including changing gene structure and dosage, alternating gene regulation and exposing recessive alleles (Henrichsen et al. 2009a; Zhang et al. 2009). Therefore, CNVs are considered a major source of genetic variation, underscoring their importance in genetic diversity and evolution. In particular, segmental duplications (SDs) were demonstrated to be one of the major catalysts and hotspots for CNV formation (Emanuel and Shaikh 2001; Sharp et al. 2005; Goidts et al. 2006; Marques-Bonet et al. 2009). Several common structural polymorphisms have been shown to be important in both normal phenotypic variability and disease susceptibility: such as CCL3L1 in HIV/AIDS, FCGR3B in glomerulonephritis, DEFB4A in Crohn's disease, C4A in lupus, and PRSS1 in pancreatitis (Gonzalez et al. 2005; Aitman et al. 2006; Fellermann et al. 2006; Le Marechal et al. 2006; Fanciulli et al. 2007; Yang et al. 2007).
A human study of the contribution of CNVs to complex phenotypes indicated that SNPs and CNVs captured ∼80% and ∼20% of the total detected genetic variation in gene expression, respectively (Stranger et al. 2007). Additionally, mouse studies provided evidence that CNVs shape tissue transcriptomes on a global scale (Cahan et al. 2009; Henrichsen et al. 2009b). Although analyses of a subset of CNVs provided evidence of linkage disequilibrium with flanking SNPs (Conrad et al. 2006; Hinds et al. 2006), a significant portion of CNVs were not easily tagged by SNPs and often fell in genomic regions (such as SDs) not well covered by SNP arrays, thus not genotyped (Estivill and Armengol 2007). Interrogation of the genome for both CNVs and SNPs, including common and rare variations, could be an effective way to elucidate the causes of complex phenotypes and disease in humans (McCarroll 2008; Manolio et al. 2009). Combining CNV and SNP data in human genome-wide association studies has associated CNVs with autism, schizophrenia, idiopathic learning disability, neuroblastoma, and severe earlier-onset obesity (Sebat et al. 2007; Cook and Scherer 2008; Bochukova et al. 2009; Diskin et al. 2009; Glessner et al. 2009; Shi et al. 2009; Stefansson et al. 2009).
Previous cattle studies have identified few local deletions ranging from 2 kb to over 200 kb (Ohba et al. 2000; Drogemuller et al. 2001; Liu et al. 2008a). A cattle CNV survey using three Holstein bulls identified 25 germline CNVs and significant amounts of CNVs waiting for discovery (Liu et al. 2008b). Additional evidence for the existence of CNV comes from bovine SNP data, where an initial screen from 556 animals of 21 cattle breeds identified 79 candidate deletion variants (Matukumalli et al. 2009). However, SNP probes on the current BovineSNP50 platform are neither dense enough nor uniformly distributed to achieve an unbiased and high-resolution cattle CNV map. Therefore, the frequency and pattern of cattle CNV events are still largely unknown as no systematic study has been reported. High-density array CGH, along with the existence of genome reference sequence, makes it possible to assess cattle CNV in a systematic, cost-effective, and high-throughput fashion. Along with a recent cattle SD study (Liu et al. 2009), we describe here the first comprehensive, systematic and genome-wide discovery and confirmation study of CNVs in the modern domesticated cattle. We identified over 200 candidate CNV regions in a panel of 90 animals from 11 Bos taurus (taurine, humpless), three Bos indicus (indicine or zebu, humped), and three composite (crosses between taurine and indicine) breeds for beef, dairy, or dual purpose. We further discuss the impact of characterizing large amounts of such variations, some of which are likely to arise independently in breeds and contribute to breed differences.
Results and Discussion
Cattle CNV discovery and distribution
Array CGH experiments were performed as previously described (Selzer et al. 2005). We used an updated version of the previously described method to identify changes in log2 signal intensity corresponding to copy number gains and losses (Olshen et al. 2004). We conservatively defined our CNV call filtering criteria to reduce false-positives called in the reference DNA self–self hybridizations (see Methods for details). By using this set of strict criteria, a total of 1041 CNVs within known chromosomes in all 90 samples passed the filters and, on average, 11.57 gain or loss events were evident in each sample (Table 1). CNVRs were determined by aggregating overlapping CNVs identified in all samples across array CGH experiments (Redon et al. 2006). Excluding chrUnAll (unassigned sequence contigs), 177 high-confidence CNVRs were detected, covering 28.1 Mb of polymorphic sequence, i.e., 1.07% of the placed chromosomes (28.1 Mb/2634.4 Mb, chr 1–29 and X in Fig. 1; Supplemental Fig. S2). On chrUnAll, we detected another 52 candidate CNVRs. Combining these two data sets resulted in a total of 229 CNVRs, corresponding to 1.57% of the bovine genome (47.7 Mb/3036.6 Mb, Supplemental Fig. S1; Supplemental Table S2). As expected, the “uncharacterized chromosome” (chrUnAll), which consists of sequence that cannot be uniquely mapped to the genome, contains the majority of predicted variable polymorphic sequence (19.6/47.7 Mb, 41.1%, see Supplemental Fig. S1; Supplemental Table S4). However, due to the lack of sequence and/or the mapping uncertainty, candidate CNVRs on chrUnAll were considered separately with caution. For example, more than 50% of the samples showed gain in few candidate regions on chrUnAll (CNVR nos. 209, 215, and 217 in Supplemental Table S2), and these regions likely reflect male vs. female differences indicating the presence of chr Y sequences in chrUnAll. Also three CNVRs (CNVR nos. 227, 228, and 229 in Supplemental Table S2) only comprised of concatenated multiple short contigs of which each was 3–20 kb in length. Since real chromosomal positions of these contigs in the genome are not known, these CNVRs are probably not real and need more investigation.
CNV events by subspecies and origins

Cattle copy number variation and segmental duplication regions display a local tandem distribution pattern. CNV regions (177 events, 28 Mb, ∼1% of the bovine genome) reported by 90 array CGH experiments are shown above the chromosomes in green (gain), red (loss), and dark blue (both). The bar height represents their frequencies: short (appeared in 1 sample), median (≥2 samples), and tall (≥5 samples). Segmental duplications (94.4 Mb, 3.1% of the bovine genome) predicted by two independent computational approaches are illustrated on the chromosomes in red (WSSD), blue (WGAC), or purple (both). The patterns are depicted for all duplications for ≥5 kb in length and ≥90% sequence identity. The gaps in the assembly are represented on the chromosomes as white ticks. For clarity, distribution patterns with the unassigned sequence contigs (chrUnAll) are shown separately in Supplemental Figure S1.
We also made CNV calls on UMD3 (Supplemental Tables S3, S4) and obtained a comparable number of CNVRs (224) after removal of the suspected candidates on chrUnAll. A simple comparison indicated that the total length of variable regions were similar with comparable statistics (4.48% difference, Supplemental Table S3). For CNVR types, count differences for each category were less than 3.59%. This is expected as both assemblies are based on the same raw whole-genome shotgun reads and the most obvious difference is that Btau_4.0 unplaced contigs were now placed on the UMD3. On the other hand, it also suggested that except for those suspected regions as described above, many of the CNVRs on Btau_4.0's chrUnAll are probably real. Since the majority of cattle genome annotations were performed on Btau_4.0, in the following analyses, we focused on further characterization of the 177 high-confidence CNV regions from Btau_4.0 known chromosomes.
These 177 CNVRs include 100 loss, 54 gain, and 23 both events (loss and gain within the same region), ranging from 18,000 to 1,261,895 bp with a mean or median of 159,031 or 89,053 bp, respectively (Table 1). Furthermore, 88 CNVRs were found in only one sample (Unique), while 89 CNVRs were found in multiple animals or breeds (Multiple) and 49 of 89 multiple events have a frequency >5% (Table 1; Supplemental Table S2). By definition, these 49 high-confidence common CNVs can be classified as potential candidate copy number polymorphisms (CNPs), if derived from the same ancestral alleles. These data sets confirm that segregating CNVs exist among 16 additional cattle breeds, which is consistent with our earlier observation of considerable genetic diversity within Holsteins (Liu et al. 2008b). In general, the number of CNVs identified in each sample is consistent with SNP estimates of breed-specific founding and effective population sizes and levels of polymorphism based on ≥50,000 SNPs (Matukumalli et al. 2009). As shown in Table 1, more CNV events were detected in indicine (17.63 per sample) and composite (15.75 per sample) than in taurine breeds (10.73 per sample), while within the taurine breeds, more CNV events were found in African breeds (11.60 per sample) than in European breeds (10.63 per sample). Although part of these differences are related to the fact that our reference sample is a Hereford cow of European origin (Dominette 01449), this observation is consistent with the subspecies divergence and supports the hypothesis of multiple independent domestications of cattle in the Fertile Crescent, Southwest Asia, and probably Africa (Troy et al. 2001; Caramelli 2006).
Cattle CNVs are distributed in a nonrandom fashion at two different levels. First, CNV content varies significantly among different chromosomes. The proportion of any given known chromosome susceptible to CNV regions varies from 0.08 to 3.49% (Supplemental Table S5). Chromosomes 5, 15, 18, 27, 29, and X show the greatest enrichment for CNV (Fig. 1; Supplemental Table S5) with twofold the variable content of the genome average excluding chrUnAll. It is interesting to note that these chromosomes also have the highest SD content (Liu et al. 2009). Furthermore, similar to the human, mouse, rat, and dog genomes, there are a greater proportion of CNVs near pericentromeric and subtelomeric regions. Excluding unmapped contigs, pericentromeric and subtelomeric regions each represent 3.4% of genomic sequence but show an enrichment of ∼2.0-fold for CNVs (both P-value < 0.001) and contain 6.7%–6.9% of all polymorphic sequence.
qPCR analysis of selected CNV regions
Quantitative PCR (qPCR) was performed using TaqMan and/or SYBR green chemistry on 65 cattle used in the array CGH experiments to further validate six selected CNV regions (see Methods). In addition to three distinct control prime pair sets, 12 independent primer pair sets were designed to target these six CNV regions (Supplemental Table S6). Five selected regions showed homology with the cattle gene clusters: olfactory receptor (CNVR28), UL16-binding protein—ULBP (CNVR56), ATP-binding cassette transporter C4 (CNVR70), bovine salivary protein 30 kDa (BSP30 in CNVR73) and zinc finger protein (CNVR104), while CNVR77 does not correspond to any known gene. As shown in Supplemental Figure S2 and Supplemental Table S7, we detected significant correlations between qPCR results and array GCH data using Monte Carlo simulations (100,000 replicates) to adjust for multiple testing. Multiple testing was completed since qPCR data from each primer pair was correlated with every probe in an array CGH gain or loss event. This was necessary because the average log2 ratio did not always reflect the magnitude of the CNV as a result of variability in hybridization intensities among probes in the segment and variability in apparent CNV boundaries among animals. At the primer pair level, 11 of the 12 (91.67%) primer pairs yielded results that correlated positively (P-value < 0.05) with array CGH hybridization data on Btau_3.1, 4.0, or both. The only primer pair that did not correlate with array CGH was ABCC4_1. Thus, at the primer pair level, qPCR data suggested a low FDR of actual calls within such regions (1/12 = 8.33%).
FISH characterization of predicted CNV regions
We experimentally validated a subset of the CNV regions by FISH (Fig. 2). A total of 41 large-insert cattle BAC clones corresponding to cattle CNV regions (>20 kb in length) were used as probes and hybridized against three Bos taurus cell lines (Angus, Hereford, and Holstein, respectively; see Supplemental Table S8). The results of all FISH experiments are available online at http://bfgl.anri.barc.usda.gov/cattleCNV/. We observed variable copy numbers either by examination of interphase or metaphase FISH for 17/41 of the probes, showing variable signal numbers either among three cells (13) or between haplotypes (4). Only one of the interchromosomal probes showed more than three distinct signals, while the majority (16/17) of intrachromosomal duplication signals were tandemly clustered. Similar to the mouse and dog genomes (She et al. 2008; Nicholas et al. 2009), these data reinforce that tandem intrachromosomal distributions of CNV are predominate in the cattle genome (Fig. 1). The basis for the remaining 24 BAC probes consistent with nonvariable regions is unknown. We note, however, that the animals for the three cell lines used in the FISH experiments were different from the animals used for array CGH, and structural polymorphism, as well as limitations of BAC-FISH to detect duplications <40 kb (especially in the case of tandem duplications) may account for differences between the array CGH predictions and FISH data.
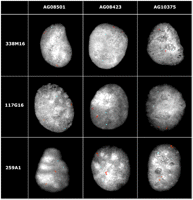
FISH confirmation. Examples of interphase two-color FISH include three BAC clones. Clones 338M16, 117G16, and 259A1 (red) were identified in CNVR31, 56 and 70, corresponding to WC1.1, ULBP17, and ULBP21, and ATP-binding cassette transporter C4, respectively. Increased signal intensity was confirmed using cohybridization with a unique control BAC clone (297K6, blue) in the same nucleus. These BACs produced variable signal count and/or intensity, 338M16: 3, 2, and 2 and 3; 117G16: 2, 2 and 3, and 2 and 259A1: 2, 3, and 3 in these three cell lines, respectively (for summary, see also Supplemental Table S8). Tandem distribution patterns were most frequently observed. The results of all FISH experiments are available online at http://bfgl.anri.barc.usda.gov/cattleCNV/.
CNVs overlap with deletion variants derived using SNP arrays
We compared our 177 CNVRs with 79 candidate deletion variants (51 unique genomic loci) reported earlier using SNP results from 556 animals of 21 breeds (Matukumalli et al. 2009). Only seven CNVRs (7/177, 3.95%) overlapped with 10 deletion variants of six loci derived from SNP results and these were verified on several common animals that were analyzed by both studies. It is expected that the majority of the variants identified in these two studies do not overlap. A similar situation was encountered in human CNV studies using the early version of SNP arrays (Eichler 2006; Matsuzaki et al. 2009). Besides differences in detection technology, we suspect that the main reasons for these discordances are due to (1) resolution differences for which array CGH arrays contain 385,000 probes with a mean interval size of 5.7 kb, while the SNP array has 54,000 probes with a mean interval size of about 50 kb; (2) sampling differences as the majority of animals in these two studies were different; and (3) genome coverage biases—by design both platforms were biased against variable genomic regions (SDs and CNVs) (Estivill and Armengol 2007). On the SNP array, there are 14 probes/Mb in cattle variable (SD and CNV) regions, compared to 21 probes/Mb in the constant regions. On the CGH arrays, there are 91–103 probes/Mb in cattle variable regions as compared to 134–136 probes/Mb in the constant regions. When we intersected these 51 deletion variants derived from SNP data with cattle SDs regions, only 23.53% of the events (12/51) overlap with cattle SDs with a overlapping space of 2,375,659 bp (5.89% of the total 40,347,982 bp). These are compared to 61.02% of the current CNVRs (108/177, see next section) that overlap with cattle SDs, corresponding to 17,937,077 bp (63.72% of the total 28,148,681 bp). We suspect the one-third reduction from 21 down to 14 probes/Mb on the SNP array could more severely interfere with CNV discovery as compared to CGH arrays. In the future, high-density unbiased CGH and SNP arrays, combined with improved CNV calling algorithms (Wang et al. 2007) could remedy this discrepancy.
CNVs overlap with segmental duplications and other genomic features
Following previous studies of other genomes, we detected a strong association between CNVs and SDs. Agreeing with cattle SDs (Liu et al. 2009), a local tandem distribution pattern is predominant in cattle CNVs (Fig. 1). It is interesting to note that about 61.02% (108/177) of high-confidence CNV regions directly overlapped with SDs. Approximately 12.25% (125/1020) of the high-confidence SDs identified by WGAC and WSSD (Liu et al. 2009) exhibit CNVs. Random simulations were repeated 1000 times and confirmed the significance of these overlaps (P-values < 0.001). We also measured overlaps using a range of genomic distances flanking CNV regions in both directions. Figure 3 displays the relationships between flanking distances and overlaps between CNV regions and SDs (either all SDs in Fig. 3A or 1020 high-confidence SDs in Fig. 3B). The colocalization remained significant (P-values < 0.001) up to 5 Mb when 177 high-confidence CNV regions were overlapped by 1020 high-confidence SDs compared with random simulations. Similar conclusions were obtained when chr X was excluded (data not shown). Agreeing with the previous cattle SD observation (Larkin et al. 2009), a strong positive correlation between CNV regions and evolutionary breakpoint regions (EBRs) was observed. Compared to the genomic averages, cattle-specific EBRs and artiodactyl-specific EBRs show 19.82% and 52.46% enrichments of CNV sequences, respectively (P-values < 0.001). We also tested the overlap between cattle CNVs and genome territories defined by ancestral and new repeat groups (Adelson et al. 2009). Similar to cattle SDs, cattle CNVs do not colocalize with either high- or low-density regions of either groups. While analysis of flanking repeats of human SDs and CNVs suggested that Alu and L1, respectively, are mainly responsible in their formation (Bailey et al. 2003; Kim et al. 2008), the coarsely mapped CNV breakpoints and the working draft nature of the bovine genome currently prevents a detailed analysis of the sequence structure at the transition regions between constant and variable sequence.
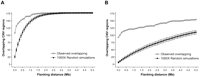
Colocalization analysis of cattle CNV regions and segmental duplications. Relationships between flanking distances and numbers of cattle CNV regions overlapped with all SDs (A) or 1020 high-confidence SD regions (B).
Although the distinctions between SDs and CNVs are not clearly defined, SDs are operationally defined as duplicated sequences (insertions) of ≥1 kb in length and ≥90% sequence identity. It is generally accepted that SDs may arise from ancient CNVs fixed in the population, providing substrates of gene and genome innovation, genomic rearrangements, and hotspots of recent CNV formation (Emanuel and Shaikh 2001; Sharp et al. 2005; Goidts et al. 2006; Marques-Bonet et al. 2009). When cattle CNV and SD data are jointly analyzed, their tandem distributions of local clusters in cattle again are reminiscent of the patterns observed in other mammals (mouse, rat, and dog), but differ from the interspersed pattern found in primate genomes. It is noted that only large CNVs (≥18 kb) were ascertained using our array CGH platform. When compared to similar studies of other mammals, the overlaps (∼50%–60%) between large CNVs and SDs are consistent among cattle, dog (Nicholas et al. 2009), and mouse (Graubert et al. 2007). A strong correlation of large CNVs and SDs in mammals supports the hypothesis that their formation mechanisms are mainly due to nonallelic homologous recombination (NAHR). On the other hand, small CNVs (<18 kb) were discovered through high-density array CGH or sequence mapping analyses (Kim et al. 2008; Cahan et al. 2009). The overlaps between small CNVs and SDs in human and mouse were significantly lower, suggesting that SDs and NAHR are less involved and other mechanisms, such as nonhomologous end-joining (NHEJ) and those proposed recently, could be more responsible (Bauters et al. 2008; Hastings et al. 2009).
Gene content of cattle CNV regions
Within known chromosomes, these 177 high-confidence CNV regions overlap with 568 Ensembl peptides, corresponding to 398 unique Ensembl genes (Table 1; Supplemental Table S9). Additionally, ∼67% (119/177) of high-confidence CNVRs completely or partially span cattle Ensembl genes. We assigned PANTHER accessions to a total of 398 overlapping genes. Statistically significant over- or underrepresentations were observed for multiple categories (Supplemental Table S10). Similar results were also obtained by using the DAVID functional annotation tool (Supplemental Table S11). This set of copy number variable genes possess a wide spectrum of molecular functions and provides a rich resource for testing hypotheses on the genetic basis of phenotypic variation within and among breeds.
Consistent with similar CNV analyses in other mammals (human, mouse, and dog), several of these CNVs, which are important in drug detoxification, defense/innate and adaptive immunity, and receptor and signal recognition, are also present in cattle. These gene families include olfactory receptors, ATP-binding cassette (ABC) transporters, Cytochrome P450, beta-defensins, T-cell receptor loci, and the bovine MHC (BoLA), which support the shared GO terms among mammals as shown in Supplemental Table S11. Conservation of CNVs across mammals suggests that selective pressure may drive acquisition or retention of specific gene dosage alterations. Since these genes or gene families have been repeatedly detected in multiple mammalian genomes, we recently surveyed the repertoires and evolutionary mechanisms of seven well-studied multimember gene families in cattle, humans, mice, and dogs (Liu et al. 2009). In summary, these multiple-member gene families normally went through the so-called “birth-and-death” evolution (Nei and Rooney 2005) in which new copies were created by gene duplication and some of them were retained in the genome for a long time as functional genes, but other copies were inactivated or eliminated from the genome. While some ancient members arose before the last common ancestor of mammals, a common theme is that new members often originated after divergence of these mammals from each other. These lineage-specific gene expansions of individual subfamilies were detected in all four species, especially in cattle and mice (see Table 2 in Liu et al. 2009). Depending on their nature (gene ancestries, structures, functions, and genomic distributions), three major evolutionary mechanisms—gene duplication, positive selection, and conversion—have shaped these gene families to different degrees.
It is intriguing to note that for many gene families that went through cattle-specific gene duplication (Liu et al. 2009), such as C-type lysozymes, BSP30A and interferon tau subfamilies, we also detected marked variation in copy number between individuals and across diverse cattle breeds (Supplemental Table S12). In the earlier cattle SD study, we detected a high level of sequence identity (median = 98.9%), which indicates that over 25% (263/1020 > 99.0%) of the bovine duplications may have occurred within the artiodactyla and Bos lineages contributing to cattle speciation and domestication (Liu et al. 2009). The current CNV survey further indicates that “birth and death” of those new copies may be still going on recently and differentially in multiple cattle breeds leading to our observations.
We also identified CNV regions that span potential cattle QTLs and human orthologous OMIM genes influencing disease susceptibility (Supplemental Table S14). For instance, multiple CNV regions directly overlap with QTLs for clinical mastitis, somatic cell count, somatic cell score, and parasite resistance. Eighteen out of 177 CNVRs correspond to known human disease genes. Other overlapping QTLs are involved in many production and reproduction traits, such as marbling score, calving ease, gestation length, pregnancy rate, and inseminations per conception. However, since cattle QTLs are less well-defined, future study is warranted.
Cattle CNV frequency differences among breeds
We generated a heat map, which revealed marked variation in copy number among cattle breeds (Supplemental Fig. S3). As discussed earlier, more CNV loci were predicted in indicine, composite, and African taurine breeds than in European taurine breeds, which is consistent with the breed divergence and history. A similar analysis of CNVRs also showed that cattle breeds tend to have similar counts generally agreeing with breed history. To highlight the potential evolutionary contributions of these CNVs to cattle breed formation and adaptation, we conservatively queried out 35 CNVRs that have high-confidence breed-specific CNV frequency differences (Supplemental Table S12). Twenty-nine of these CNVRs correspond to annotated genes or gene families, while many of them are also known in other mammals to interact with environments, some of them are known to be important in cattle adaptation including SCP2 (Liu et al. 2009), ULBP (Larson et al. 2006), and WC1.1 (Herzig and Baldwin 2009). To our knowledge, this is the first systematic report on breed-specific copy number differences in cattle. Based on these differences of CNV frequency among cattle breeds, we hypothesize that some cattle CNVs are likely to arise independently in breeds and contribute to breed differences and therefore are related to the breed formation and adaptation.
We performed a detailed study of CNVR56 corresponding to a known ULBP gene cluster on chr 9 (Fig. 4; Supplemental Table S13). After mapping of the ULBP major cluster (Larson et al. 2006), we found within this region ULBP21 and ULBP2 are potential functional copies, while other copies are either partially overlapped (ULBP17) or pseudogenes (Supplemental Fig. S4). Our PCR and FISH results further confirmed the presence and copy numbers of these variations. Cattle ULBP1 and ULBP2 genes encode members of the MHC Class I superfamily. In human, ULBP1 and ULBP2 interact with the KLRK1 (NKG2D) receptor to activate effector cells in the immune system, a critical resistance factor for cytomegalovirus infection. Considering the overall average ratio between gain and loss is nearly 1:2 (54:100), it is a dramatic contrast to note the gain and loss ratio in this CNV region is 18:5, indicating an adaptative/positive selection for or a relaxed purification against increasing copies of ULBP. Our data indicate that the Limousin breed has the highest gain events (50%, 5/10, including Limousin3, 4, 5, 7, and 9), while the Angus breed has the highest loss events (27%, 2/11, Angus4 and 7). It is also interesting to note Limousin9 has both loss and gain events within this ULBP gene cluster.
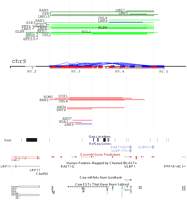
A detailed analysis of CNVR56 corresponding to the major ULBP gene cluster (chr9:90,150,000–90,550,000). On the chromosome, cattle SDs are predicted by WSSD (brown) and WGAC (blue and red represent intra- and interchromosomal WGAC duplications). Above chromosome are gain events (green = three copies and dark green = four copies), while below are loss events (one copy) of corresponding regions arranged vertically according to their relative log2 ratios from the chromosomal baseline. Limousin9 displays both loss and gain events (labeled as blue) within this region. The UCSC gene and expression tracks are shown at the bottom. Five light blue vertical lines represent potential breakpoint regions. ANG, Angus; BAN, Brangus; CHL, Charolais; GIR, Gir; GLB, Gelbvieh; GZR, Guzerat; HFD, Hereford; HOL, Holstein; LMS, Limousin; NDA, N'Dama; RAN, Red Angus; ROM, Romosinuano; and SMT, Simmental.
Within a cattle pedigree and a number of parent-offspring trios, selected deletion variants have been shown to be stably inherited across generations in cattle population (Liu et al. 2008a; Matukumalli et al. 2009). To rule out this possibility, we examined the pedigree among these affected animals and did not find any significant existing relationship. Furthermore, based on the event length, we detected at least five distinct breakpoint regions, which have been repeatedly used by direct visualizations, suggesting at least four distinct event types in Limousin and two distinct event types in Angus (Fig. 4). Assuming the reference genome as two copies, four states were detected (one, two, three, and four copies). The combining evidence strongly suggests that in Limousin, all CNVs in CNVR56 were probably not derived from a single inherited ancient event, but instead it is more plausible that multiple recurrent, discrete de novo gain or loss events of distinct origins occurred in this CNV region. Additional evidences from other breeds also support this notion. When we mapped the gene structures on this CNVR (Supplemental Fig. S4) we found that the righthand part of Figure 4, roughly corresponding to ULBP2, was amplified up to four copies in Limousins 3 and 7, while the lefthand part, roughly corresponding to ULBP21, was decreased down to one copy, producing a hemizygous state in Limousin9. Besides the ULBP gene clusters, Supplemental Table S12 lists additional CNVRs with breed differences such as CNVR28 (olfactory receptor) in Angus, Red Angus, and Holstein (see also Liu et al. 2008a) and CNVR12 (SCP2) in Gelbvieh and Red Angus. These examples provide proof of principle that some CNVs may underlie many phenotypic differences between cattle breeds. Obviously, these observations, while interesting, require additional genomic structural and functional studies to better delimit the relationship between gene copy number (genotype) and variation in breed traits (phenotype).
Recent breed-specific positive selection may elevate population differentiation. To explore breed differentiation at all CNVs, we performed a VST analysis, as described previously (Redon et al. 2006). We identified 687 array CGH probes with levels of breed differentiation suggestive of breed-specific selective pressures at a false discovery rate (FDR) of 20% (Supplemental Table S15). Among them, 130 probes have an FDR value of <10% and 45 probes of <5%. The distribution of population variable probes in cattle appears to be similar to human (Redon et al. 2006). These probes overlap eight CNVR regions, including the above-discussed gene clusters: CNVR28 (olfactory receptor), CNVR70 (ABCC4), CNVRs 24 and 31 (WC1.1), and CNVR94 (complement factor HF1) (Supplemental Table S14). Since not all regions that have been under recent positive selection exhibit elevated population differentiation, we also overlapped our CNVRs with two sets of genomic regions under positive or balance selection detected by iHS and FST using SNP data (The Bovine HapMap Consortium 2009; Flori et al. 2009). Eight significant overlaps were noted including CNVR1 (similar to adenylate cyclase 5), 11 (similar to guanylate binding protein 4), 17, 24 (GIMAP GTPase), 36, 75, 76 (similar to brain adenylate cyclase 8 isoforms), and 82 (Supplemental Table S14). However, the observed differences between breed variations could be caused by both selection and genetic drift due to genetic bottlenecks for some breeds. It needs further confirmation using a larger sample size.
Conclusions
In this project, we employed an integrated approach combining array CGH screens, qPCR confirmations and FISH verifications to study cattle CNVs. The extent of this variation, and some of the gene classes affected, are similar to other mammals. We have presented the frequency, pattern, and potential impact of such cattle-specific CNVs. Most cattle CNVs affect genes for specific biological functions, such as immunity, lactation, reproduction, and rumination, and are thus likely to be functional. We identified 35 CNV regions that may be breed-differential or breed-specific. These CNV differences among cattle breeds could be due to altered metabolic and immune requirements due to microbial fermentation in the rumen, the herd environment, and the reproductive strategy of cattle through human selection. Our combined findings reveal that some cattle CNVs are likely to arise independently in breeds and contribute to breed differences, thus associated with cattle domestication and breed formation. Our CNV results provide insight into mechanisms of bovine genome evolution and generate a valuable resource for cattle genomics research. This high-quality cattle CNV map fills the gaps left out by the current SNP-based genome-wide association and selection studies. A more comprehensive appreciation of the full dimension of bovine genetic variation may unravel the genetic basis for the further genetic improvement of milk and beef production.
It is unlikely that this initial cattle CNV list reported here is complete as the CGH arrays were designed using only one reference genome. As a result, sequences absent in Dominette and present in other animals cannot be ascertained. With the costs of genome sequencing dropping dramatically by using next-generation sequencing, emerging high-quality cattle genomic sequence will soon facilitate the application of this direct sequence comparison strategy. Approaches such as paired-end sequence mapping strategy have yielded massive numbers of new genomic structural variations at high resolution that will improve future CNV research (Tuzun et al. 2005; Korbel et al. 2007).
Methods
Selection of cattle breeds and animals:
Using the pedigree and the breed phylogeny trees constructed for cattle mitochondria DNA and ∼35,000 SNPs (Troy et al. 2001; The Bovine HapMap Consortium 2009), breeds and individuals were selected as divergent as possible to represent the current North American cattle population. Due to artificial insemination, which is commonly used in the cattle industry, we expected a reduced diversity in the commercial cattle populations. We determined the relationship matrix among our samples: The highest average relationship is less than 0.08 and the highest average inbreeding coefficient is less than 0.05. The chosen 17 breeds and their origins and features are summarized in Supplemental Table S1 including 11 Bos taurus breeds: Angus, Bonsmara, Charolais, Gelbvieh, Hereford, Holstein, Limousin, N'Dama, Red Angus, Romosinuano, and Simmental; three Bos indicus zebu breeds: Brahman, Gir, and Guzerat; and three composite or cross breeds: Beefmaster, Brangus, and Santa Gertrudis. Genomic DNA samples were purified from semen, whole blood, and ear notch as described (Sonstegard et al. 2000). All DNA samples were analyzed by spectrophotometry and agarose gel electrophoresis.
Control hybridizations and somatic variations
We conducted control hybridizations using the sequenced Hereford cow L1 Dominette 01449 and its sire L1 Domino 99375 (American Hereford Association registration nos. 42190680 and 41170496, respectively). We performed array CGH using Dominette's blood DNA (female) and Domino's semen DNA (male) including female self-to-self, male self-to-self, and female vs. male to evaluate baseline variations. To rule out somatic variations, control hybridizations were also conducted among different tissues of the same donors (Dominette's skin vs. whole blood in one pairwise comparison and Domino's semen, skin, and whole blood in all three pairwise combinations). Under our conservative calling criteria (see below), all self-to-self and self-tissue hybridizations showed no detectable false-positive or somatic variations. We then fixed the blood DNA samples from Dominette (Dt blood) as the reference sample in all hybridization experiments.
Array CGH
Whole-genome CGH arrays contain ∼385,000 oligonucleotide probes (http://www.nimblegen.com) that were designed and fabricated on a single slide to provide an evenly distributed coverage with an average interval of ∼6 kb using either Btau_3.1 or Btau_4.0 genome assemblies (The Bovine Genome Sequencing and Analysis Consortium 2009). These types of arrays utilize synthetic probes 50–75 bp in length with similar melting temperatures and do not require sample amplification or reduced representation. Standard genomic DNA labeling (Cy3 for samples and Cy5 for references), and hybridizations, array scanning, data normalization, and segmentation were performed as described earlier (Olshen et al. 2004; Selzer et al. 2005; Graubert et al. 2007).
The CNVs were represented by gains and losses of normalized fluorescence intensities relative to the reference. The high-confidence calls are filtered and merged according to the similar criteria as described previously, i.e., we merged overlapping CNV coordinates across hybridizations to form unique CNV regions using the 40% overlapping threshold as described previously (Redon et al. 2006; Graubert et al. 2007). For cattle CNV calling, we first migrated the probes from Btau_3.1 to Btau_4.0 and made calls on Btau_4.0. We then tested a series of log2 ratio shift and affected neighboring probe counts and their impact on the FDR in the self–self-control hybridizations. We selected a set of conservative calling criteria for the final set of high-confidence CNVs, requiring alternations of 0.5 log2 ratios over five neighboring probes (0.5_5), under which no false-positive was found for self–self-control hybridizations. Therefore, the arrays have a resolution of ∼24 kb. For chr X, the baselines were shifted to negative because all test samples are bulls (males, one chr X) and the reference sample was a cow (female, two chr X). We also compared other settings including 0.5_3, 0.3_5, and 0.3_3. For example, the 0.5_3 setting yielded 44 more regions and ∼2 Mb more sequence (273 regions covering 49,651,971 bp vs. 229 regions covering 47,725,392 bp), but produced one positive in self–self-control hybridizations.
Three distinct CNV regions on chrUnAll that were supported by the majority of male cattle were labeled to indicate their potential chr Y origins: no. 209, chrUnAll:130,132,172–130,488,613 (55/90); no. 215, chrUnAll:164,064,002–164,699,372 (50/90); and no. 217, chrUnAll:173,853,000–173,925,000 (84/90) (Supplemental Table S2). Thus, these observations suggest that the FDR among the set of predicted CNVs is low (∼1.3%), although technical issues, such as sequence divergence of individual cattle relative to the reference genome sequence or heterogeneity in DNA quality among samples makes it difficult to precisely quantify the FDR. We also migrated probes from Btau_3.1 and Btau_4.0 to UMD3 using liftOver and repeated the entire calling analyses for all 90 hybridizations to ensure consistency in calls (Supplemental Table S3). Because of the strict filtering criteria, a noticeable false-negative rate was expected.
qPCR confirmation
We performed qPCR using the relative comparative threshold cycle (CT) method to confirm copy number changes detected by array CGH. These include tests on 65 cattle and six CNV regions using TaqMan and/or SYBR green chemistry on a MJ Chromo4 RT-PCR machine (Bio-Rad) as recommended by the manufacturers. Two distinct PCR primers and/or probes were designed to target each CNV region using Primer3 (Supplemental Table S4). CT values in triplicate were averaged and normalized against the control gene for each assay. Assuming that there were two copies of DNA in the control regions, the relative copy number for each test region was calculated as 2(1+ddCT). The significance (5% and 1% level) of correlation between PCR results and array CGH data was tested using 10,000 times Monte Carlo simulations after adjusting for multiple testing as previously described (Wain et al. 2009). Briefly, we correlated the qPCR results for each primer pair separately with log2 ratios within the candidate CNV region due to the uncertainty of the CNV breakpoints. To overcome the multiple testing problem, for every primer pair and build combination, we generated a vector of random normal variables (length equal to the number of animals, mean = 0 and variance = 1). We then ran the correlation between that random vector at each position separately. We obtained a correlation at each position for each simulation of qPCR data. Next, we sorted these correlations from smallest to largest. We repeated this 100,000 times for each probe. This resulted in a 100,000 by (number of probes in segment) matrix where the column is rank of the correlation and row is the replicate. We then estimated the 0.5, 2.5, 97.5, and 99.5 percentiles for each rank. Correlation estimates exceeding 2.5 and 97.5 percentiles are significant at the 5% level, while correlation estimates exceeding 0.5 and 99.5 percentiles are significant at the 1% level.
FISH validation
FISH experiments were performed as described previously (Ventura et al. 2003; Liu et al. 2009). Forty-one cattle BAC clones (CHORI-240) were selected with large (≥20 kb) copy number variable regions as determined by array CGH. Both interphase and metaphase nuclei were prepared using three cell lines (AG08501: Hereford male smooth muscle cell; AG08423: Angus female fibroblast; and AG10375: Holstein male fibroblast from Coriell Cell Repositories). A single BAC clone (297K6) was used as control in each FISH experiment. Differentially labeled test and control BAC clones were cohybridized to one slide. To determine the copy number of the test BAC, we calculated the ratio between the number of signals of the test BAC and the number of signals of the control BAC. We counted 40–50 nuclei for each slide and reported their averages. Metaphase nuclei were examined to identify chromosomal origins of FISH signals. More intense FISH signals, which localized to a single site, were subsequently examined by interphase nuclei. Interphase analyses were controlled for replication by comparing cells at both G1 and G2 stages of arrest.
Cattle CNV distribution and association with segmental duplications and other features
We investigated the genomic distribution of CNVRs by testing the hypothesis that pericentromeric and subtelomeric regions were enriched for CNVs, as we did previously for cattle SDs. All predicted variable bases that overlap these regions were totaled and χ2 tests were used to test the null hypothesis of no enrichment as previously described (Liu et al. 2009). Association between CNVs and SDs was tested by 1000 times random simulations by selecting valid genomic segments from the length distribution of 177 high-confidence CNVs and determining if the segments overlapped at least one SD. Additional genomic features are obtained from public databases listed in website references.
Gene content
Gene content of cattle CNV regions was assessed using Ensembl genes (ftp://ftp.ensembl.org/pub/current_fasta/bos_taurus/pep/), the Glean consensus gene set, cattle RefSeq and in silico mapped human RefSeq (the UCSC Genome Browser, http://genome.ucsc.edu/). Intersections between CNV region coordinates and exon positions were compared using MySQL queries. We obtained a catalog of all bovine peptides from Ensembl. This yielded 26,271 peptides, 568 of which overlap with predicted 177 high-confidence CNV regions, and corresponded to 398 unique Ensembl genes. Using the PANTHER classification system, we tested the hypothesis that the PANTHER molecular function, biological process, and pathway terms were under- or overrepresented in CNV regions after Bonferroni corrections (Nicholas et al. 2009). It is worth noting that a portion of the genes in the bovine genome have not been annotated or have been annotated with unknown function, which may influence the outcome of this analysis. However, another independent Gene Ontology and pathway analysis (DAVID, http://david.abcc.ncifcrf.gov/) was based on human–cow alignment net and overlapping human orthologous gene annotation using the UCSC Genome Browser. DAVID (Supplemental Table S11) also produced results similar to PANTHER (Supplemental Table S10).
Population genetic and statistical analyses
Breed-specific CNVs were estimated using the VST analysis as described earlier (Redon et al. 2006). Briefly, VST is calculated by considering (VT − VS)/VT, where VT is the variance in log2 ratios apparent among all unrelated individuals and VS is the average variance within each breed, weighted for breed size. Nominal P-values were computed for VST using analysis of variance with breed as a one-way classification. FDR for VST was estimated as described previously (Benjamini and Yekutieli, 2001).
Acknowledgments
We thank members of the Bovine HapMap Consortium for sharing their samples. We thank D. Hebert, T. Brown, W. Gang, and H. Preston for technical assistance. This work was supported in part by NRI grant no. 2007-35205-17869 from USDA CSREES (now NIFA) and Project 1265-31000-098-00 from USDA-ARS. E.E.E. is supported by NIH grant GM058815 and is an investigator of the Howard Hughes Medical Institute.
Author contributions: G.E.L. and J.W.K. conceived and designed the experiments. Y.H., B.Z., L.J., A.M., L.K.M., and J.S. performed in silico prediction and computational analyses. T.G.M., R.W.L., M.F.C., M.E.D., G.L., A.C., and M.V. performed qPCR and FISH confirmation. L.J.A., C.P.V.T., T.S.S., L.C.C., L.C.A.R., D.N., T.P.L.S., and L.C.G. collected samples. G.E.L., T.G.M., J.W.K., and E.E.E. wrote the paper.
Footnotes
-
↵13 Corresponding authors.
E-mail John.Keele{at}ars.usda.gov; fax (402) 762-4155.
E-mail George.Liu{at}ars.usda.gov; fax (301) 504-8414.
-
[Supplemental material is available online at http://www.genome.org. The array CGH data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE19866.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.105403.110.
-
- Received January 19, 2010.
- Accepted March 4, 2010.








