
Dynamic reprogramming of transcription factors to and from the subtelomere
- 1 Division of Biological Sciences, University of California San Diego, La Jolla, California 92093, USA;
- 2 UCSD Moores Cancer Center, University of California San Diego, La Jolla, California 92093, USA;
- 3 Department of Bioengineering, University of California San Diego, La Jolla, California 92093, USA
Abstract
Transcription factors are most commonly thought of as proteins that regulate expression of specific genes, independently of the order of those genes along the chromosome. By screening genome-wide chromatin immunoprecipitation (ChIP) profiles in yeast, we find that more than 10% of DNA-binding transcription factors concentrate at the subtelomeric regions near to chromosome ends. None of the proteins identified were previously implicated in regulation at telomeres, yet genomic and proteomic studies reveal that a subset of factors show many interactions with established telomere binding complexes. For many factors, the subtelomeric binding pattern is dynamic and undergoes flux toward or away from the telomere as physiological conditions shift. We find that subtelomeric binding is dependent on environmental conditions and correlates with the induction of gene expression in response to stress. Taken together, these results underscore the importance of genome structure in understanding the regulatory dynamics of transcriptional networks.
It is becoming increasingly evident that gene order is not random (Hurst et al. 2004; Kosak and Groudine 2004). Expression profiling of diverse eukaryotic species has revealed that coexpressed genes are often clustered along the chromosome (Cohen et al. 2000; Versteeg et al. 2003; Su et al. 2004; Pauli et al. 2006) and that such clusters of genes function to varying degrees in the same metabolic pathways (Lee and Sonnhammer 2003). In yeast, for example, adjacent genes are coregulated during the cell cycle (Cho et al. 1998) or in response to changing growth conditions (Kruglyak and Tang 2000). In multicellular eukaryotes such as flies and humans, extended tracts of coexpression have been observed encompassing up to 30 genes (Caron et al. 2001; Spellman and Rubin 2002).
These effects of gene order on expression, collectively known as “position effects,” are controlled by a variety of mechanisms that are still incompletely understood (Hurst et al. 2004). A substantial body of work in the yeast Saccharomyces cerevisiae, in which strong position effects are observed near chromosome ends, shows that position effects depend on epigenetic factors such as the state of surrounding chromatin and the spatial compartmentalization of the nucleus (Mondoux and Zakian 2006). These epigenetic factors are, in part, assembled by DNA-binding transcription factors and chromatin modifying proteins (Grewal and Jia 2007; Sexton et al. 2007). At least one transcription factor (Rap1p) and several chromatin modifiers (including Hda1p and the Sir silencing complex) appear to bind in a position-specific manner at the distal tips of chromosomes—that is, at the telomeres—or across multiple genes in the genomic region adjacent to the telomere known as the subtelomere (Gottschling et al. 1990; Robyr et al. 2002; Rusche et al. 2003). These proteins, however, do not completely explain the observed expression patterns of subtelomeric genes, many of which are thought to function in different stress response pathways (Wyrick et al. 1999; Ai et al. 2002; Robyr et al. 2002).
Given that subtelomeres contain clusters of functionally related genes, and given that only a few telomere- and subtelomere-associated transcription factors have been found, one might expect that additional such factors might remain to be discovered. In particular, we sought to address a number of questions related to transcriptional regulation at chromosome ends: How many factors are used to regulate the telomere or subtelomere? Do they localize exclusively to the ends of chromosomes or are they also found at other sites throughout the genome? Given that subtelomeric genes are regulated dynamically in response to stress, are these dynamics also reflected in transcription factor binding?
Here, we address these questions by computationally screening the wealth of available chromatin immunoprecipitation (ChIP) data in yeast for evidence of position-specific binding. We find that a surprising number (more than 10%) of all profiled yeast DNA-binding transcription factors display a marked preference for binding genomic locations within 25 kb of the telomere and that much of this position-specific binding is responsive to changes in physiological conditions. We also assay the phenotypes of single and double deletions of these transcription factors in response to physiological challenges. None of these factors have been previously known to have any correlation with genome position, but we find that seven are highly connected via physical interactions with proteins with known telomere functions. Taken together, our findings suggest that genome position effects involve not only coexpression of neighboring genes along a genome but are also evident in the architecture and dynamics of entire transcriptional regulatory networks.
Results
Discovery of a large family of subtelomere binding transcription factors
As the initial basis for our analysis, we used the compendium of transcription factor (TF) binding profiles published by Harbison et al. (2004). These binding profiles were gathered using the technique of ChIP followed by microarray analysis (ChIP-chip) for each of 203 S. cerevisiae TFs. All TFs were assayed in yeast grown in rich medium, and a subset of these TFs was also assayed under stress conditions.
We scored each TF using a quantitative measure of telomere-proximal binding which we call its telomere distance profile (TDP). We computed the TDP for a TF by measuring the distance to the closest telomere for every target sequence reported to be bound by that TF, resulting in a distribution of distances. Then, we compared each TDP to a background TDP consisting of all yeast genes (Fig. 1A; see Methods).
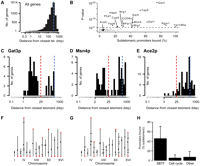
Transcription factors that preferentially bind sequences at subtelomeres. (A) Background telomere distance profile (TDP) for all yeast promoters. (B) Overview of TDPs for rich-medium promoter-binding profiles (Harbison et al. 2004) compared against the background distribution. Each dot represents data for one TF, its significance score on the y-axis (the P-value of a one-sided Kolmogorov-Smirnov test) versus the percentage of bound promoters that are subtelomeric on the x-axis. Subtelomere binding transcription factors (SBTFs) having bimodal TDPs more significant than the P-value threshold of 0.001 are labeled and indicated in black. (C–E) TDPs for Gat3p (C), the statistically most significant SBTF; Msn4p (D), a SBTF that is a master regulator of stress response genes; and Ace2p (E), a cell cycle regulator that is representative of TFs having a TDP similar to the background. Blue broken lines correspond to the blue broken line in A. The red broken line indicates the 25-kb cutoff distance used to categorize subtelomeric genes. (F,G) Plots showing the location of promoters bound on the 16 yeast chromosomes by Gat3p (F) and Msn4p (G). Red diamonds indicate binding events located within 25 kb of a telomere. Black ticks indicate nontelomeric binding events. Small black dots mark the centromere on each chromosome. (H) The average percent of subtelomeric promoters bound by three groups of TFs: SBTFs, cell cycle TFs (those annotated with the “cell cycle” Gene Ontology term GO:0007049), and the remaining TFs. Error bars, SD.
Among the TF binding profiles from yeast grown in rich medium, 17 TFs had a TDP that was significantly different from the background distribution according to a Kolmogorov-Smirnov (KS) test at P < 0.001 (Fig. 1B). This P-value threshold corresponds to a false discovery rate (FDR) (Storey and Tibshirani 2003) of ∼1%, meaning that none of the TDPs identified are expected to be false positives (Supplemental Fig. 1).
Fifteen of these significant TDPs were distinctly bimodal, with an unusually high number of target sequences located within 25 kb of the closest telomere (Fig. 1C,D,F,G). This bimodality was not simply due to low gene density 25 kb from the telomere, since the background distribution was not depleted for genes in this region (Fig. 1A). In S. cerevisiae, the subtelomere is postulated to extend roughly 25 kb inward from each telomere (Louis 1995). Thus, we named these 15 factors SBTFs (subtelomere binding transcription factors).
Next, we analyzed the 84 TF binding profiles that had been reported under various stress conditions such as rapamycin, butanol, or hydrogen peroxide (Harbison et al. 2004). Eleven SBTFs were identified that showed a subtelomeric binding preference under stress. Conversely, a number of SBTFs that had been identified in rich medium were found to lose their subtelomeric binding preferences in alternative conditions (Fig. 2).
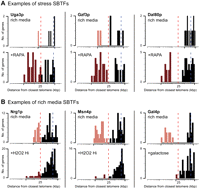
Example SBTFs that show dynamic binding preferences as a function of growth or stress condition. (A) SBTFs with TDPs that are significant only in a stress condition. TDPs are shown for each SBTF in two conditions: rich medium (top) and a stress condition (bottom). Genes considered subtelomeric are colored light red in rich-medium binding profiles or dark red in stress profiles. (B) SBTFs that show subtelomeric preference in rich medium. Stress conditions are abbreviated: RAPA (100 nM rapamycin), H2O2 Hi (4 mM hydrogen peroxide), and galactose (2% in YEP medium). Red broken lines, 25 kb. Blue broken lines, peak of background distribution as indicated in Figure 1A.
In total, this raised the number to 22 SBTFs identified (Fig. 3A): seven TFs for which the subtelomeric binding preference was specific to a stress condition; six TFs for which it was specific to rich media; four TFs for which subtelomeric binding was observed under both stress and rich media; and five TFs for which no stress binding data were available. In one case, a TF (Nrg1p) showed different subtelomere binding behaviors in two stress conditions, but was only counted once (that is, as subtelomeric binding in stress and rich media). The importance of the subtelomere per se is underscored by our observation that SBTFs bind relatively few genes in the genomic regions immediately adjacent to the subtelomere, 25–50 kb from chromosome ends (Supplemental Fig. 2). It is perhaps intriguing that 11% of S. cerevisiae TFs (22/203) display a binding preference for subtelomeric genes, which themselves only comprise about 6% of the genome.
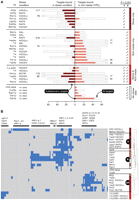
Subtelomeric binding preference is dynamic and distributed into distinct clusters. (A) Comparison of stress versus rich-medium SBTF promoter binding profiles. Dark and light horizontal red bars indicate the number of subtelomeric targets bound. Open bars indicate the total number of targets bound. Check marks on right indicate whether the SBTF shows a significant (P < 0.001) subtelomeric preference in the stress condition (S) or rich (R) medium. (B) Hierarchical clustering of the 125 subtelomeric genes (columns) bound by one or more SBTFs (rows). Blue indicates binding. Colored bars at right correspond to the dynamic binding behaviors in A: SBTF displays a subtelomeric preference in stress only (S; dark red), rich media only (R; light red), or both stress and rich media (SR; bright red). Stress conditions are abbreviated: Acid (succinic acid at pH 4); BUT90 (1% butanol); GAL (2% galactose); RAFF (2% raffinose); H2O2Lo (0.4 mM hydrogen peroxide); H2O2Hi (4 mM hydrogen peroxide); RAPA (100 nM rapamycin); SM (0.2 mg/mL sulfometuron methyl, an inhibitor of amino acid biosynthesis).
SBTFs regulate stress and carbon metabolism in three broad clusters
Roughly one-third of the SBTFs we identified had been previously associated with cellular stress responses (i.e., Msn4p, Pdr1p, Phd1p, Rox1p, Xbp1p, Yap5p, Yap6p; see Table 1). SBTFs also included factors mediating glucose and nitrogen repression (Mig1p, Dal80p, Gzf3p, Nrg1p, Uga3p) as well as growth on alternative carbon sources (Gal4p, Mal33p, Hap4p) and metal uptake (Aft2p, Cup9p).
Summary of subtelomere binding transcription factors
In contrast, TFs associated with most other cellular programs, such as the cell-cycle regulator Ace2p (Fig. 1E), had TDPs that closely matched the background distribution (Fig. 1H). Five SBTFs (Gat3p, Dat1p, Rgm1p, Yjl206c, Ypr196w) were poorly characterized or of unknown function, while SBTFs such as Gal4p, Msn4p, and Pdr1p were among the most well studied transcription factors in yeast. Their identification as subtelomeric binding factors is, to our knowledge, novel, but it is in concordance with their known roles in regulating stress or metabolic genes (Mefford and Trask 2002).
Hierarchical clustering of SBTF binding profiles indicated that SBTFs bound at least three distinct classes of subtelomeric genes (Fig. 3B; Supplemental Fig. 3). Stress-only SBTFs largely targeted alcohol dehydrogenases (AAD3 and ADH7) along with YRF genes, a family of putative helicases located within the subtelomeric Y′ repeated element, which may function in telomere maintenance when telomerase is absent (Yamada et al. 1998). Rich-media SBTFs bound YRF genes as well as members of the COS gene family, which are widely conserved and may function in salt resistance (Mitsui et al. 2004) and the unfolded protein response (Spode et al. 2002) but are otherwise generally uncharacterized.
Intriguingly, SBTFs identified in both stress and rich media conditions bound a completely distinct set of stress responsive promoters upstream of hexose transporters (HXT), flocculation genes (FLO, FSP2), and a sorbitol dehydrogenase (SOR1). Notable exceptions to these trends included Mig1p, a SBTF only in rich media but which bound targets in the “stress and rich media” cluster; Yap6p, which bound different sets of subtelomeric genes in low versus high levels of hydrogen peroxide; and three SBTFs (Aft2p, Mal33p, Yjl206c) that did not strongly cluster with other factors.
SBTFs do not behave like known telomere-binding complexes or transcription factors but do interact with these proteins
The telomere, as opposed to the subtelomere, is the target of several extensively studied protein complexes. The function of these complexes includes telomere replication, chromosome end protection, and transcriptional silencing. To assess whether SBTFs have been linked to any of these functions, we examined protein complexes (Krogan et al. 2006; Supplemental Fig. 4), recent literature reviews (Lundblad 2006; Mondoux and Zakian 2006), and results from a genetic screen for telomere length mutants (Askree et al. 2004). None indicated that SBTFs play known roles near the telomere (Supplemental Table 1). However, we also analyzed all of the protein interactions from the BioGRID database (Stark et al. 2006) and did find that SBTFs, as a group, were statistically enriched (P = 0.02) for TFs highly connected via physical interactions to telomere proteins (Supplemental Fig. 5; Supplemental Table 2). These physical interactions with proteins having known telomere functions reinforce the finding that SBTFs preferentially bind near chromosome ends.
We next manually inspected the TDPs of TFs known to influence telomere length and silencing (Askree et al. 2004; Lundblad 2006; Mondoux and Zakian 2006). The most extensively studied such factor, Rap1p, binds to repeated sequences (C1–3A) that are found at all chromosome ends and plays roles in telomere silencing and length control (Lundblad 2006). We note that in our computational screen Rap1p was identified among the top subtelomere binding factors (P = 0.011; Table 1) but did not meet our stringent P-value cutoff. This is consistent with previous reports that found >80% of the target sequences bound by Rap1p lie outside the subtelomere in the promoters of many essential genes (Lieb et al. 2001). Another transcription factor, Met18p, was identified in a screen for mutants that affect telomere length (Askree et al. 2004) and just missed our stringent P-value cutoff. These results suggest that our analytical method is indeed capable of recovering known proteins that function at chromosome ends.
Evaluating the effects of cross-hybridization
Since subtelomeric DNA is highly repetitive (Louis 1995), it is plausible that a single bound subtelomeric promoter might cross-hybridize to multiple probes on the microarray used to assay promoter binding. To control for this possibility, we correlated SBTF binding profiles with the presence of 10 non-open reading frame (ORF) sequence features annotated by the Saccharomyces Genome Database, which include the X, Y′, telomeric repeat, and autonomic replicating sequence elements (Supplemental Table 3). The binding profiles for two SBTFs, Yap5p and Msn4p, were correlated with the Y′ element (Bonferroni-corrected Pearson P < 0.01 with r = 0.75 and r = 0.73, respectively), but no other significant correlations were found, suggesting that subtelomeric binding cannot be attributed to the known repeats alone.
To further estimate the possible effects of cross-hybridization, we applied TDP analyses to a filtered data set in which binding targets with similar promoter sequences had been removed. Promoter similarity was determined by whether the polymerase chain reactions—used to generate the promoter DNA spotted on the ChIP-chip microarrays—were predicted to amplify more than two different genomic sequences (Harbison et al. 2004). Most SBTFs remained significant in this data set (15 of 22). We also assessed the similarity of all yeast promoters using comprehensive pairwise BLAST comparisons. Similar promoter sequences were not typically bound by the same TF—a pattern consistent with minor effects of cross-hybridization (Supplemental Fig. 6). Taken together, these analyses suggest that the discovery of SBTFs cannot be attributable to the repetitive nature of subtelomeric DNA.
Six SBTFs target heterochromatin domains that are derepressed during growth on alternative carbon sources
Next, we sought to explore whether SBTFs might be associated with Hda1p, the only known regulator that specifically targets the subtelomere. Hda1p is a histone deacetylase that establishes HDA1-associated subtelomeric (HAST) domains that repress about 40% of all subtelomeric genes (Robyr et al. 2002). It is plausible that SBTFs could help establish, maintain, or relieve repression of genes in HAST domains.
We examined the clusters of subtelomeric genes bound by SBTFs (Fig. 3B) and found a significant number of HAST genes in cluster “SR,” the stress-responsive genes bound by Mig1p, Nrg1p, Phd1p, and Yap6p in hydrogen peroxide, butanol, and rich conditions (Fig. 4A; binomial test, P < 0.01; Supplemental Table 5). These same HAST genes were also bound by Xbp1p in hydrogen peroxide. Interestingly, the other SBTF in the SR cluster, Yjl206cp, also bound HAST genes, albeit a different set.

Binding profiles, expression, and growth phenotypes link SBTFs to Hda1p. (A) SBTFs that have a significant percentage of targets in HAST domains (P < 0.01). Exact numbers of targets are above each bar. (B–D) Box-and-whisker plots of gene expression changes in an hda1Δ strain compared with wild type (data from Bernstein et al. 2002) for targets bound by Yap6p (P = 10−8) (B); Phd1p (P = 0.00006) (C); Yap1p (statistically unaffected) (D). (E) YAP6 and PHD1 display a condition-specific genetic interaction during growth on non-glucose carbon sources. Fivefold serial dilutions were grown on synthetic complete (SC) medium supplemented with 2% of one of the following carbon sources: glucose, galactose, ethanol, or glycerol. Controls are BY4741, the parent strain used to create gene deletions, and gal4Δ, which does not grow on galactose.
We found that the subtelomeric genes bound by TFs in cluster SR were specifically up-regulated in an hda1Δ deletion strain (Bernstein et al. 2002) (Fig. 4B–D; Supplemental Fig. 7; KS test, P < 0.001), consistent with Hda1p's function as a repressor. However, SBTFs in this cluster did not appear to be required for establishing or maintaining repression because individually deleting each TF (Hu et al. 2007) does not affect the expression of many subtelomeric targets in rich medium (Supplemental Fig. 8).
Taken together, these findings suggest that SBTFs in cluster SR perhaps do not function at the subtelomere by differential binding but instead are modulated by other regulatory mechanisms such as post-translational modification. Alternatively, it is possible that they do function by differential binding but only in conditions other than those profiled to date.
Since genes in HAST domains function during growth on alternative carbon sources (Robyr et al. 2002), we tested the SBTF deletion strains yap6Δ, phd1Δ, nrg1Δ, and yjl206cΔ for growth defects on solid medium supplemented with glucose, fructose, galactose, lactose, ethanol, maltose, or sucrose using a series of dilution assays. Although growth defects were not observed for any single mutant, a yap6Δ phd1Δ double mutant exhibited a clear growth defect in galactose, ethanol, and glycerol (Fig. 4E).
This interaction likely depends on subtelomeric genes, as Yap6p and Phd1p bind seven common subtelomeric targets (Supplemental Table 6) including the hexose transporter-like genes HXT9, HXT15, and HXT16. The gene HXT15 was previously found to have higher expression levels during growth on ethanol and glycerol (Greatrix and van Vuuren 2006).
In summary, we have linked six SBTFs to the chromatin-modifying enzyme Hda1p, which mediates Sir-independent silencing at the subtelomere. Two of these SBTFs, Yap6p and Phd1p, jointly contribute to growth on alternative carbon sources, a role consistent with existing models of Hda1p function.
Dynamic subtelomeric binding is correlated with gene expression
Given that subtelomeric genes are typically repressed (Wyrick et al. 1999), a SBTF that preferentially binds the subtelomere in a stress condition (cluster S in Fig. 3B) might function as an activator if its targets are up-regulated in the same stress. Alternatively, a SBTF that moves away from the subtelomere in response to stress (cluster R) might function as a repressor if its targets are subsequently up-regulated in the same condition.
To distinguish between these hypotheses, we analyzed published gene expression profiles (Gasch et al. 2000) to identify SBTFs whose subtelomeric target genes had condition-specific regulatory behaviors (McCord et al. 2007; Lee et al. 2008) (see Methods). We discovered a strong correlation between the binding and upregulation of targets of Aft2p in response to oxidative stress. This correlation was particularly striking for the 11 members of the subtelomeric PAU gene family that are bound by Aft2p under mild hydrogen peroxide treatment (Fig. 5A), although the profiles of some family members might be affected by microarray cross-hybridization with other PAU-family genes since these sequences are over 80% similar to PAU1 at the nucleotide level. PAU genes and Aft2p have each been implicated in oxidative stress resistance (Rachidi et al. 2000; Blaiseau et al. 2001), but our results provide the first evidence that Aft2p up-regulates PAU genes.
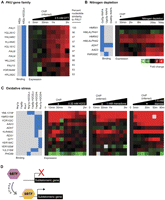
Dynamic binding and expression profiles suggest models of SBTF function and link SBTFs to poorly characterized genes at subtelomeres. TF binding profiles were matched to expression profiles gathered under similar environmental perturbations. (A) Aft2p binds upstream of 12 subtelomeric PAU genes under oxidative stress conditions (blue boxes). Heatmap shows induction of the PAU genes in oxidative stress conditions. (B) In the presence of rapamycin, which simulates nitrogen depletion by antagonizing the TOR kinases (Magasanik and Kaiser 2002), Gzf3p, Uga3p, and Dal80p bind upstream of genes that are induced under conditions of nitrogen limitation. (C) Genes targeted by Nrg1p, Yap6p, and/or Rox1p under hydrogen peroxide but not in untreated conditions are up-regulated in response to three oxidative stress agents (hydrogen peroxide, menadione, and diamide). For A–C, the asterisk (*) indicates the approximate time that the binding data were collected. (D) SBTFs that display a preference for the subtelomere only in stress conditions may be positive regulators of gene expression (see text). Expression heatmaps all use scale shown in B.
The other stress-only and rich-media-only SBTFs bound two overlapping sets of genes (Fig. 3B). These included 30 genes similar to the YRF family that we found to be largely unresponsive in stress conditions (Supplemental Fig. 9). However, excluding the YRF-like genes, three stress-dependent SBTFs (Gzf3p, Uga3p, Dal80p) displayed behaviors consistent with functions as activators in conditions of nitrogen depletion (Fig. 5B).
The expression analysis also identified a set of subtelomeric genes that were targeted by Yap6p, Nrg1p, or Rox1p under hydrogen peroxide but not in untreated conditions and that were up-regulated in response to a broad array of oxidative stress agents (Fig. 5C). For these targets, the three SBTFs appear to behave like “stress-only” SBTFs, although Yap6p and Nrg1p had been classified as “stress-and-rich-media” factors based on their global binding patterns in Figure 3A. Intriguingly, many of the putative stress-regulated targets of these SBTFs are poorly characterized and unnamed, although YML131W is similar in sequence to oxidoreductases (Hong et al. 2008). In contrast to the above evidence for SBTFs as transcriptional activators, we did not find any strong evidence for SBTFs as repressors of gene expression.
Discussion
We have identified 22 yeast transcription factors that display a clear binding preference for subtelomeric regions (Fig. 1; Table 1). Several factors are found to be highly connected via physical interactions to genes encoding proteins with known telomeric functions (Supplemental Fig. 5). The majority of binding patterns are dynamic, such that the factor is concentrated at the subtelomere only under certain conditions (Figs. 2, 3). This finding, combined with our analysis of stress-induced expression profiles, suggests that most stress-only SBTFs (cluster S) are activators of subtelomeric genes in response to stress (modeled in Fig. 5D) and may relocalize to different target genes under other conditions.
Previous observations that subtelomeric genes may function in stress conditions (for review, see Mondoux and Zakian 2006) are supported by Gene Ontology (GO) enrichment analysis of subtelomeric genes, which reveals subtelomeric concentrations of ion and sugar transporters as well as genes involved in alternative carbon metabolism (data not shown). Given these observations and the nonrandom distribution of genes (Hurst et al. 2004), one might expect that a TF involved in regulating transport or alternative carbon metabolism might display a subtelomeric binding preference. On the other hand, among the 14 TFs found to bind upstream of five or more genes annotated as ion transporter or alternative carbon metabolism genes, only six are SBTFs (43%), indicating that the function of target genes alone is not sufficient to predict that a TF will show a subtelomeric binding preference.
The identification of SBTFs adds yet another chapter to a growing body of evidence suggesting strong mechanistic links between genome organization, nuclear architecture, and gene expression (Schneider and Grosschedl 2007; Sexton et al. 2007). One model for the function of rich-media SBTFs is that they reside in transcriptional repressive foci within the nucleus. Indeed, foci at the nuclear periphery have been found to contain telomeres (Klein et al. 1992) along with other regions of silent heterochromatin (Feuerbach et al. 2002; Komili and Silver 2008). Alternatively, in a process termed “reverse recruitment,” some genes appear to be recruited to sites of active transcription tethered to nuclear pore complexes (Sexton et al. 2007; Komili and Silver 2008). Since Rap1p has been implicated in this process (Casolari et al. 2004), it is tempting to ascribe a similar role to SBTFs. Under this model, the condition-dependent binding seen in Figure 3 would be interpreted not as SBTFs themselves “moving toward” or “away” from the subtelomere but rather as different subtelomeric genes moving in or out of sites of gene activation or repression at the nuclear periphery.
One must also consider the possibility that a SBTF might be neither an activator nor a repressor. Instead, it may be sequestered to subtelomeres without effecting changes in subtelomeric gene expression as a means of holding it in reserve from other genomic locations. There may be roles for SBTFs at the telomere that are dependent on sequestration as suggested for Rap1p (Marcand et al. 1996; Lieb et al. 2001) and shown for the telomeric Ku proteins that become mobilized to sites of DNA damage to facilitate repair (Bertuch and Lundblad 2003). Additionally, since telomere ends have been suggested to loop back to subtelomeric regions (Bystricky et al. 2005), the SBTFs identified in this study could have a role in that process.
As greater numbers of binding profiles are generated in yeast and other species, it may be fruitful to screen those data for unexpected binding preferences at the subtelomere as well as more generally at other genomic regions. For example, just such a study was published while this paper was in review (Janga et al. 2008). In addition, genome-wide binding analysis of Sgo1p and Rec8p, two cohesin proteins that function in chromosome segregation, has been used to show that they localize to a 50-kb region around the centromere (Kiburz et al. 2005). Our work demonstrates that many other such analyses may be productive, and it provides an example of transcription factor binding patterns that reflect genome organization.
Further work will be required to determine the extent to which condition-specific targeting of the subtelomere is a conserved regulatory strategy. Orthologs for all SBTFs have been identified in one or more other yeast species, and seven SBTFs appear to be widely conserved across fungi, invertebrates, fish, and mammals (Supplemental Fig. 10). Moreover, SBTFs that cluster together by binding profile (Fig. 3B) also appear to have roughly similar patterns of conservation across species. Intriguingly, paralogs of most SBTFs were themselves not found to be SBTFs, with the exception of Dal80p and its paralog Gzf3p. This suggests there may be selective pressure that maintains the subtelomeric binding pattern of one paralog but that redundancy is typically not necessary. It is tempting to speculate that SBTFs and their dynamic localization may contribute to the evolutionary plasticity that has previously only been attributed to the subtelomeric genes themselves (Mefford and Trask 2002; Louis and Vershinin 2005).
Methods
Computational screen for SBTFs
SBTFs were screened from published genome-wide TF binding data (Harbison et al. 2004). A P-value threshold of 0.001 was used to identify the set of gene promoters putatively bound by each TF in a particular environmental condition (either rich medium or one of 12 additional stress or nutritional conditions).
To compute the distance from each gene to the closest telomere, we first computed midg, the midpoint of the starting and ending chromosomal coordinates for gene g, which were downloaded from the Saccharomyces Genome Database (http://www.yeastgenome.org). Then, the distance to the closest telomere is dg = min(midg, LengthChrg – midg), where LengthChrg is the length of the chromosome on which g is located.
The distribution of distances for all genes targeted by a TF in a particular binding experiment was defined to be the TDP. Using R (http://www.r-project.org), a one-sided KS test was used to compare the TDP for each binding experiment to a background distribution of the TDP for all yeast genes. To estimate the FDR, the set of all KS P-values was used as input to the Q-value software (Storey and Tibshirani 2003).
Statistically significant TDPs were identified at P ≤ 0.001, corresponding to a FDR of ∼1%. Subtelomeric genes were defined to be those with dg ≤ 25000. Hierarchical clustering of SBTF binding profiles was performed using Cluster 3.0 (de Hoon et al. 2004) and visualized using Java TreeView (Saldanha 2004). To simplify data visualization, clustering was limited to subtelomeric genes bound by at least one SBTF.
Analysis of protein complexes and interactions involving SBTFs
A comprehensive set of yeast protein–protein interactions was obtained from the BioGRID database (Stark et al. 2006) version 2.0.40 (May 2008). Interactions that connected a SBTF to any known telomere-related gene were identified and then manually inspected using Cytoscape (Shannon et al. 2003). Physical interactions were classified as those in BioGRID labeled: Affinity Capture, Co-crystal Structure, Co-fractionation, Co-purification, FRET, Far Western, Protein–peptide, Protein–RNA, Reconstituted Complex, or Two-hybrid. Genetic interactions were classified as those in BioGRID labeled: Dosage Growth Defect, Dosage Lethality, Dosage Rescue, Phenotypic Enhancement, Phenotypic Suppression, Synthetic Growth Defect, Synthetic Lethality, or Synthetic Rescue.
A compendium of 547 nonoverlapping yeast protein complexes (Krogan et al. 2006) was checked to see whether any complex contained one or more SBTFs and any of the known telomere-related genes listed in Supplemental Table 1. Telomere-related genes were obtained from two sources: (1) any gene mentioned in recent reviews of yeast telomeres (Lundblad 2006) or the telomere position effect (Mondoux and Zakian 2006) or discovered in genetic screens for telomere length mutants (Askree et al. 2004); and (2) any gene annotated in the GO database under the categories “telomere organization and biogenesis” (GO: 0032200) or “chromatin-silencing-at-telomere” (GO: 0006348). In total, 426 telomere-related genes were identified, 321 from GO analyses and an additional 105 from literature reviews.
Analysis of repetitive element binding by SBTFs
Yeast genome sequence features were downloaded from the Saccharomyces Genome Database (http://downloads.yeastgenome.org/chromosomal_feature/SGD_features.tab). All features labeled ORF were discarded, leaving 10 different types of non-ORF features. Next, we determined the presence or absence of one or more occurrences of each feature on the 32 S. cerevisiae chromosome arms. We then determined whether each SBTF bound at least one promoter on the 32 chromosome arms. These analyses generated two sets of vectors consisting of 32 elements each: one set corresponding to SBTF binding and another set corresponding to sequence features. Using the Pearson correlation test in R, the similarity of each of the binding vectors was compared with each of the 10 non-ORF feature vectors. P-values were adjusted for multiple hypotheses testing using the Bonferroni correction.
Screening SBTF binding profiles for correlation with hda1Δ sensitivity
We used a method by Steinfeld et al. (2007) to identify TF and chromatin modifying enzyme (CM) pairs that function in concert. Briefly, gene expression profiles of CM deletions were analyzed to determine whether the targets of a TF, identified by ChIP-chip, were preferentially affected. The rationale is that if a TF and CM function together, then deletion of the CM should preferentially affect the genes targeted by that TF.
We extended this approach by reasoning that if a TF and CM function together to specifically regulate the subtelomeric targets of a TF, then those subtelomeric targets should be preferentially expressed compared with the nonsubtelomeric targets in a CM deletion. We applied this approach to hda1Δ expression data (Bernstein et al. 2002) and the sets of subtelomeric and nonsubtelomeric genes targeted by each SBTF. As in Steinfeld et al. (2007), we used the KS test to assess the statistical significance of the difference in expression between the bound versus unbound subtelomeric targets for each SBTF. The significance threshold was a Bonferroni-corrected P-value less than 0.001.
Dilution assays for growth of SBTF deletions on alternative carbon sources
Single deletion yeast strains were obtained from the Yeast Deletion Collection (Open Biosystems). Double deletions were a gift from S. Bandyopadhyay (University of California San Diego) in the Ideker laboratory and were constructed as described previously (Schuldiner et al. 2006). Individual colonies growing on YPD agar plates were picked and cultured overnight to saturation in 2 mL of YPD. Overnight cultures were diluted with sterile water in a 96-well microtiter plate in six fivefold serial dilutions with a starting OD600 between 2 and 3. A 48-pin replica pinner was used to spot ∼5 μL of each dilution onto solid agar plates of synthetic complete medium containing 2% of one of the following carbon sources: glucose, raffinose, glycerol, ethanol, galactose, fructose, maltose, or lactose. Plates were incubated at 30°C for at least 3 d. Initial screening results were confirmed with repeated experiments.
Inferring SBTF modes of action by integrated analysis of matched binding and expression profiles
Gene expression experiments from Gasch et al. (2000) were matched to TF binding experiments (Harbison et al. 2004) performed under similar environmental perturbations. The set of gene targets bound by SBTFs in the presence of hydrogen peroxide was matched to expression perturbations caused by the oxidative agents menadione, DTT, diamide, or hydrogen peroxide. Binding profiles from cells treated with rapamycin were matched to expression profiles labeled “Nitrogen depletion.” Rich-media binding profiles were matched to expression profiles labeled “YPD,” “diauxic shift,” and “steady state expression.”
Matched sets of binding and expression that passed both of the following criteria were identified for in-depth manual inspection: (1) the expression of the subtelomeric targets bound by each SBTF was different than the unbound subtelomeric genes, as assessed by the KS test at P < 0.05; (2) the SBTF bound the differentially expressed targets only in stress conditions, or the SBTF bound these genes only in rich medium. For the most part, we did not observe a global increase in expression that was specific to genes bound by SBTFs (Supplemental Fig. 11). Further investigation revealed that the global nonresponsiveness of the stress SBTFs was due to the fact that the YRF and COS genes, which comprised a large fraction of the genes bound, were not differentially expressed. After excluding the YRF and COS genes from our analysis, we observed the binding patterns reported in Figure 5 and the main text. To identify YRF-like genes, the protein sequence encoded by YRF1-1 was used as an input to BLASTP, and matches were selected at an E-value cutoff of 10−10.
Identification of SBTF orthologs
Fungal orthologs of each S. cerevisiae gene were extracted from the ‘Pillars.tab’ file downloaded from the Yeast Gene Order Browser version 2.0 (Byrne and Wolfe 2005; http://wolfe.gen.tcd.ie/ygob/data/Version1.0_Nature-2006/Pillars.tab). Orthologs in other species were extracted from raw datafiles downloaded from version 6.0 of Inparanoid (Remm et al. 2001) (http://inparanoid.sbc.su.se/download/old_versions/6.0/sqltables/). The reported number of orthologs for an ORF in another species is the total number of distinct orthologs for that ORF and for the S. cerevisiae paralogs of that ORF defined in the YGOB.
Acknowledgments
We thank S. Bandyopadhyay for providing the double deletion strains used in this project; S. Jacobson for assisting with dilution assays; S. Bandyopadhyay, D. Gottschling, S. Jacobson, J. Karlseder, T. Ravasi, K. Tan, B. Trask, C. Workman, and C.H. Yeang for critical comments on the manuscript. This work was funded by grant ES14811 to T.I. from the NIEHS and by grant GM56469 to L.P. from NIH. T.I. is a David and Lucille Packard Fellow.
Footnotes
-
↵4 Corresponding author.
E-mail trey{at}bioeng.ucsd.edu; fax (858) 534-5722.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.084178.108.
-
- Received August 6, 2008.
- Accepted February 9, 2009.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








