
Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion
Abstract
Methylation of cytosines (mC) is essential for epigenetic gene regulation in plants and mammals. Aberrant mC patterns are associated with heritable developmental abnormalities in plants and with cancer in mammals. We have developed a genome-wide DNA methylation profiling technology employing a novel amplification step for DNA subjected to bisulfite-mediated cytosine conversion. The methylation patterns detected are not only consistent with previous results obtained with mC immunoprecipitation (mCIP) techniques, but also demonstrated improved resolution and sensitivity. The technology, named BiMP (for Bisulfite Methylation Profiling), is more cost-effective than mCIP and requires as little as 100 ng of Arabidopsis DNA.
DNA methylation plays a central role in ensuring accurate epigenetic inheritance (Bird 2002). In plants and mammals, DNA methylation is necessary for normal development (Finnegan et al. 2000; Morgan et al. 2005), parental imprinting (Baroux et al. 2002; Morgan et al. 2005), and for mammalian X chromosome inactivation (Reik and Lewis 2005). Methyl groups are found at cytosines; in mammals, they are normally at those preceding guanines (mCG). In plants, in addition to mCG, mCs are present at CNG and asymmetric CHH sequences (N = A, C, G, T; H = A, T, C). In humans, aberrant mCG distributions have been implicated in aging and disease development, most notably cancer (Esteller 2007). Given the central importance of DNA methylation, genome-wide determination of precise mC distributions is of interest.
The resolution of current DNA methylation profiling methodologies is limited by the length of DNA fragments used as query probes on microarrays. For example, the use of methylation-sensitive restriction endonucleases requires DNA fractions of 1- to 4-kb for hybridizing to microarrays (Lippman et al. 2004; Tran et al. 2005). Other techniques, such as affinity chromatography with methyl-binding domain proteins (Jorgensen et al. 2006) or anti-mC antibodies to enrich for methylated DNA (named mCIP or MeDIP) (Weber et al. 2005; Keshet et al. 2006; Zhang et al. 2006) also employ inherently large probe fragments that limit resolution (Mockler et al. 2005). Moreover, large fragments also increase the likelihood of co-precipitating adjacent, unmethylated DNA, causing false-positive signals, especially on high-density oligonucleotide arrays. Finally, these approaches require relatively large amounts of input DNA (2–20 μg); thus, their application to small tissue samples remains difficult.
To increase resolution, bisulfite treatment of DNA, in which cytosines are converted to uracil but mC remains unmodified (Frommer et al. 1992), represents a promising approach (Mockler et al. 2005). Bisulfite-mediated cytosine conversion and subsequent PCR creates C-to-T transitions, which can be detected by sequencing, or possibly, on oligonucleotide arrays, analogous to single nucleotide polymorphisms (Borevitz et al. 2003; Singer et al. 2006). Indeed, probes derived from bisulfite-converted DNA of a hypermethylated locus in human tumors proportionally increased signal intensity in a methylation-dependent manner when hybridized to methylation-specific oligonucleotide arrays (Gitan et al. 2002). However, bisulfite methods have not been previously applied for genome-wide DNA methylation profiling. This is likely due to DNA fragmentation during bisulfite treatment, precluding the preparation of microarray probes representing all chromosomal loci equally.
Here, we prepared genomic probes suitable for high-density oligonucleotide tiling arrays using a novel technique that reduces the amplification bias for bisulfite-treated DNA. We performed methylation profiling with the same Arabidopsis strains and arrays previously used for mC immunoprecipitation (mCIP) experiments (Zhang et al. 2006). Our results not only agreed with previous mC immunoprecipitation (mCIP) data (Zhang et al. 2006), but also identified additional methylation polymorphisms due to increased resolution.
Results
A novel random amplification method for BiMP
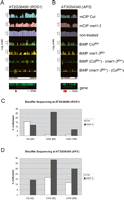
Prolonged bisulfite treatments to obtain complete cytosine conversion lead to severe DNA fragmentation (Warnecke et al. 2002). Methods to protect DNA during bisulfite reactions improve recovery of fully converted DNA (Raizis et al. 1995; Olek et al. 1996). Despite using a similar method (see Methods), when using a standard whole-genome amplification technique, referred to as “sixN” (Iyer et al. 2001; Lippman et al. 2005), we observed variations in the representation of different chromosomal sequences for bisulfite-converted DNA (Fig. 1). Moreover, the degree of amplification bias at some loci varied strongly between replicates. To circumvent this problem, we reduced the length of the random primers from 6-mers to 4-mers to favor the priming of smaller DNA fragments. With these modified primers, herein referred to as “fourN,” we observed improved uniformity of amplification at randomly chosen single-copy chromosomal loci (exemplified by the AT3G08650 locus, Fig. 1A; Supplemental Fig. 1A). The improved fidelity of the “fourN” amplification was also observed for repetitive sequences, whose specific detection by dot blot hybridizations reflected an efficient bisulfite conversion (Fig. 1B). These results suggested that bisulfite conversion followed by the “fourN” amplification produced material suitable to prepare probes for Affymetrix microarrays. For bisulfite conversion, we used 2 μg of genomic DNA, but we found that 100 ng of bisulfite-converted DNA was sufficient to perform a “fourN” amplification reaction producing enough DNA (>9 μg) to prepare probes for a high-density microarray. Bisulfite methylation profiling (BiMP) can, therefore, be achieved with relatively small amounts of starting material. During the probe preparation, a modified fragmentation treatment was used (see Methods) to produce fragment sizes averaging 60–80 bp (Supplemental Fig. 1B), supporting the optimal resolution of the high-density tiling array profiles.

Post-amplification quality assessment following random amplification methods used for Bisulfite Methylation Profiling (BiMP). (A) Assessment for amplification bias using locus-specific PCR amplification at the AT3G08650 locus. For each DNA sample, two random amplification methods were performed, “sixN” and “fourN” (as indicated on the left) and assayed for the presence of the target template (see Supplemental Fig. 1A). The second- (2nd) and fourth-generation (4th) homozygotes of the met1-3 mutant are indicated. Col is indicated as “na” (not applicable). DNA sample treatment is indicated as either bisulfite-converted (+) or non-treated (−). Each lane corresponds to an equal volume of PCR reaction (10 μL). DNA was separated in a 1% agarose gel and stained with ethidium bromide. The PCR-positive control (pos control) was amplified from genomic Col DNA. The template-free controls (no-template control) correspond to the Sequenase elongation (1st), random amplification (2nd), and PCR (PCR) steps (see Methods). The gel images have been manipulated to create a contiguous lane order. (B) Evaluation of repeat representation in randomly amplified reaction products using dot blot hybridizations. Non-treated and bisulfite-converted Columbia DNA samples were amplified by the methods indicated (left) and hybridized to a macroarray (see Methods). The macroarray was spotted with 200 ng of PCR amplicons derived from Col DNA with (+) or without (−) bisulfite-conversion from different genomic repeat sequences (indicated above and described in the Methods). Each column of the macroarray was spotted with two technical replicates, observed as rows on the macroarray. Rectangular borders were added to the image to separate the bisulfite-converted (+) from the nonconverted (−) probes.
Reproducibility of the BiMP analysis
We performed hybridizations on the Affymetrix 1.0R Arabidopsis tiling array using bisulfite-converted DNA subjected to “fourN” amplification originating from plants depleted of METHYLTRANSFERASE 1 (MET1) and the corresponding wild type, herein named met1-3BS+ and ColBS+, respectively. MET1 is the primary maintenance methyltransferase responsible for propagating mCGs (Saze et al. 2003). We first assessed the technical reproducibility of the “fourN” amplification method by comparing hybridization results derived from identical DNA samples that were subdivided for independent amplification, labeling, and hybridization. The technical replicates had correlation coefficients of 0.98 and 0.90 for ColBS+ and met1-3BS+, respectively (Fig. 2A,B). The lower correlation coefficients for met1-3BS+ may be explained by the inherent variation in methylation patterns among individuals observed in this strain (Mathieu et al. 2007). Also, increased variance was observed for both at low hybridization signal intensities, a common observation for microarrays utilized for transcription profiling (Zhu and Wang 2000) and mCIP (Keshet et al. 2006).

Assessment of the technical reproducibility observed for the BiMP technology. A scatter plot comparison of two technically replicated hybridization datasets (one hybridization per axis) with all signal intensities presented (>3.2 × 106 probe pairs, log2 scale). From one bisulfite-converted sample, two DNA aliquots (100 ng) were independently amplified, fragmented, labeled, and hybridized (see Methods). The correlation coefficient (r) was calculated using TAS (Affymetrix), and the plot was drawn in the R statistical environment (http://www.r-project.org). (A) Col bisulfite-converted DNA, (B) met1-3 bisulfite-converted DNA.
Next, we assessed the biological reproducibility by comparing three hybridization datasets for ColBS+ and met1-3BS+, where plant material, DNA isolation, bisulfite treatment, “fourN” amplification, labeling, and hybridizations were all performed independently (see Methods). The correlation coefficients were 0.96 and 0.92 for the ColBS+ and met1-3BS+ datasets, respectively (Supplemental Table 1). These results were compared with the mCIP DNA methylation profiling performed on the same Affymetrix platform for the same Arabidopsis strains (Zhang et al. 2006; datasets available at http://www.ncbi.nlm.nih.gov/projects/geo/, accession no. GSE5094). The calculated mCIP correlation coefficients were 0.94 and 0.92 for the wild type and met1-3, respectively (Supplemental Table 1), and thus were similar to the BiMP values.
To control for the genome-wide uniformity of the “fourN” amplification and the probe behavior of each array feature, we introduced an additional reference of nonconverted Arabidopsis DNA amplified by the “fourN” method. The global signal distribution of this reference control (Supplemental Fig. 2) indicated a low failure rate of “fourN” amplification. Noticeably, in comparison to the reference control, hybridization signals for ColBS+ and met1-3BS+ were significantly decreased, likely reflecting cytosine conversions caused by the bisulfite conversion (Supplemental Fig. 2). However, many array features retained high signal intensities, possibly reflecting probes resistant to conversion due to DNA methylation (Supplemental Fig. 2). Given the previous reports of changes in DNA methylation patterns due to the met1-3 mutation (Soppe et al. 2002; Tariq et al. 2003; Mathieu et al. 2007) and the available mCIP data (Zhang et al. 2006), we assessed whether the hybridization signals correctly reflected mC distributions in the wild-type and met1-3 DNA.
Validation of the BiMP method
Analysis of methylation patterns at the chromosome level revealed that BiMP-derived profiles were consistent with previous data. For example, in the wild type, strong signals were observed at the heavily methylated heterochromatic, centromeric, and pericentromeric regions compared with the generally euchromatic chromosomal arms (Fig. 3A; Supplemental Fig. 3). Furthermore, comparison of the ColBS+ with the met1-3BS+ DNA displayed decreased signal intensity for met1-3, reflecting its genome-wide hypomethylation, notably at specific regions such as the heterochromatic knob on chromosome 4 (Fig. 3A; Lippman et al. 2004). This is further illustrated by detected changes in methylation distributions at an 80-kb centromeric/pericentromeric region of chromosome 1 (Fig. 3B). Like the 180-bp centromeric repeats, which are known to be extensively and stably demethylated in met1-3 (Mathieu et al. 2007), this region displayed such profiles using BiMP (Fig. 3B). Thus, we conclude that the BiMP results are not arbitrary. Additionally, comparison of the BiMP met1-3BS+ with the mCIP met1-3 at this region indicated increased resolution, as illustrated by the detection of relatively short stretches of DNA with different methylation patterns in the wild type and met1-3 that were less obvious in the mCIP datasets (Fig. 3B).
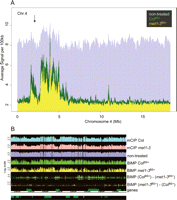
Bisulfite methylation profiling results visualized at the chromosomal level: chromosome 4 (A) and a pericentromeric region of chromosome 1 (B). (A) Graphs represent the average signal intensity per 100 kb (see Methods). (Green) ColBS+ hybridization, (yellow) met1-3BS+ hybridization, (purple) nontreated DNA hybridization. (X-axis) Physical length of chromosome 4 (NCBI Arabidopsis genome assembly version 5), (arrow) heterochromatic “knob” region. (B) Average hybridization signal intensity profiles across the pericentromeric region of chromosome 1 (chr1:15,520,000–15,600,000). Each tier represents a graph of the hybridization profile corresponding to each dataset (labeled at the right). The hybridization graphs were displayed in the range 0–11 (log2 scale). The difference graphs, BiMP (ColBS+) − (met1-3BS+) and BiMP (met1-3BS+) − (ColBS+), compare the BiMP profiles. The signal intensity differences above the applied cut-off (>4.0, horizontal line) are indicated for significantly hypomethylated intervals (green boxes) and hypermethylated intervals (yellow boxes) in met1-3BS+ relative to ColBS+.
Given the reproducibility of the BiMP datasets and the methylation analyses at heterochromatic regions, we examined single-copy loci known to change methylation levels in met1-3 mutants. For example, the FWA and SUPERMAN (SUP) genes are hypomethylated and hypermethylated, respectively, in met1-3 relative to the wild type (Chan et al. 2005). BiMP analysis of the diagnostic tandem repeats within the 5′ region of FWA clearly showed the loss of methylation in met1-3 (Fig. 4A). Conversely, the 5′ region of the SUP locus revealed hypermethylation in met1-3 (Fig. 4B).

Analysis of BiMP results at known DNA methylation targets in Arabidopsis. The graphs were designed as in Fig. 3B. (A) AT4G25530 (FWA) locus. (Red box) Region encoding the methylated repeat sequences in the wild type (Soppe et al. 2000). (B) AT3G23130 (SUP) locus. (Red box) Region that was bisulfite sequenced (see Supplemental Fig. 5).
When mCIP and BiMP feature-level signal intensities were each processed using Affymetrix’s Tiling Analysis Software (TAS) (see Methods), the improved sensitivity and resolution of BiMP over mCIP was confirmed (Figs. 3B, 4). Previously, the mCIP results (available at http://epigenomics.mcdb.ucla.edu/DNAmeth/ and http://signal.salk.edu/cgi-bin/methylome) were analyzed using a two-state hidden Markov model (HMM) based on probe-level t statistics (Zhang et al. 2006). However, this approach is likely to miss short intervals as differentially methylated, for example at the SUP locus (Supplemental Fig. 4). This is likely due to the observed high mCIP signal intensity in Columbia across the 5′ genic region that could mask adjacent local changes (Fig. 4B) or because the levels were classified as indifferent from the average global distribution. Detection of methylation at SUP in Columbia was unexpected, considering prior bisulfite sequencing analyses at this locus (Jacobsen and Meyerowitz 1997; Jacobsen et al. 2000; Kishimoto et al. 2001; Lindroth et al. 2001). Since previous analyses had been performed in different Arabidopsis accessions, we determined methylation levels in the Col accession with bisulfite sequencing at this locus (Fig. 4B; Supplemental Fig. 5). Our results confirmed the presence of methylation as detected by BiMP.
In addition, we compared the mCIP and BiMP signal intensities at eight loci known to be methylated in the wild type (Bao et al. 2004; Henderson et al. 2006; Zhang et al. 2006). The BiMP results for all agreed with the previous data (Supplemental Figs. 6, 7). Recently, DNA hypermethylation has been reported at the BONSAI locus in a DNA methylation-deficient Arabidopsis strain (Saze and Kakutani 2007). In the BiMP datasets, we observed hypermethylation within this gene in met1-3, accompanied by met1-3 hypomethylation in the LINE retrotransposon flanking the BONSAI locus (Supplemental Fig. 8). Again, the BiMP results were confirmed by restriction analysis and bisulfite sequencing (H. Saze, pers. comm.).
Since the methylation polymorphisms at FWA and SUP are best documented and unequivocally detected by BiMP, we used them to reveal novel DNA methylation polymorphisms across the genome. We assigned a positive cutoff level at 4.0 (representing a 16-fold signal difference) with a sliding window of 161 bp, roughly the sequence length per nucleosome. Under these conditions, ∼4% of the methylation intensity differences between the entries were classified as significant. These methylation polymorphisms consisted of 26,777 hypomethylated and 15,184 hypermethylated intervals, representing ∼2.7% (3,249,039 bp) and 1.3% (1,533,464 bp) of the array (1.19 Mb), respectively. Considering that total wild-type levels of mC ranged from 4%–6% (Leutwiler et al. 1984), the observed changes are rather drastic. On average, the regions that gained or lost methylation were 121 and 101 bp in length, respectively, indicating that methylation changes predominantly occur within relatively short intervals that would be difficult to visualize with mCIP given its maximum resolution estimated at 400 bp (Zhang et al. 2006). We analyzed these hyper- and hypomethylated intervals for the relative proportions of sequence motifs containing cytosines. Notably, asymmetric CHH sites were more frequent in the hypermethylated compared with the hypomethylated intervals (Supplemental Tables 2, 3). This supports and significantly extends our previous observation that following the loss of mCGs in the met1-3 mutant, there is a genome-wide gain of DNA methylation with a preference for cytosines at CHH sequences (Mathieu et al. 2007).
To validate novel methylation polymorphisms revealed by BiMP, we used conventional bisulfite sequencing. Increased methylation in met1-3 was observed at the REPRESSOR OF SILENCING 1 (ROS1) and APETALA 3 (AP3) genes (Fig. 5A,B; Supplemental Fig. 9), and the sequencing results supported the BiMP data (Fig. 5C,D). Importantly, methylation in met1-3 also included CG sites that were not methylated in the wild type (Fig. 5D; Supplemental Fig. 10). At the assayed AP3 region, the signal difference was slightly below the chosen cutoff, suggesting that our analysis was a conservative estimate of differentially methylated regions. Methylation profiles between mCIP and BiMP at additional loci were identified (Supplemental Fig. 11) and examined by bisulfite sequencing. The BiMP results were supported (Supplemental Fig. 12). However, clonal analysis of bisulfite-converted DNA, although widely used for assessing DNA methylation, cannot be considered as quantitative as other, more expensive techniques (Dupont et al. 2004). Moreover, DNA methylation distributions and levels in met1-3 plants can vary among individuals (Mathieu et al. 2007); thus, some of the observed differences between the BiMP and mCIP studies could be inherent to the experimental material.
Validation of novel methylation polymorphisms detected using BiMP. Graphs were prepared as in Fig. 3B. (A) AT2G36490 (ROS1), (B) AT3G54340 (AP3). (Red boxes) Regions that were bisulfite sequenced. (C,D) Bisulfite sequencing at the AT2G36490 (ROS1) (C) and AT3G54340 (AP3) (D) loci. The DNA methylation level (% methylated, Y-axis) was determined by bisulfite sequencing and presented for each different sequence motif: CG, CNG, and CHH (H = A, T, C) (see Methods). The total number of cytosines analyzed per sequence motif is provided within the parentheses.
Discussion
The major accomplishment of the BiMP technology was to reduce amplification bias in the bisulfite-converted DNA, thus allowing preparation of genome-wide probes for high-density tiling arrays. Amplification uniformity was reflected by the reproducibility of methylation profiles when technical and biological replicates were compared. Moreover, comparisons between the wild type and met1-3 mutant revealed clear DNA methylation differences, which were validated by bisulfite sequencing at several selected and randomly chosen loci and by comparison to previously available genomic methylation profiles. Both validation procedures confirmed the reliability of the BiMP technology.
Importantly, comparisons of the BiMP data with the previous mCIP results for the same Arabidopsis strains and microarray platform (Zhang et al. 2006) showed not only a good concordance of methylation profiles, but also demonstrated that the resolution of profiles obtained through BiMP appears to be considerably higher. This allowed detection of methylation polymorphisms in short DNA intervals, including regions where hyper- and hypomethylated were adjacent. Therefore, BiMP is likely to reveal specific, localized changes in DNA methylation characteristics that are likely to be undetected by mCIP.
Our BiMP analyses add a genomic perspective to the previous understanding of the Arabidopsis response to the loss of CG methylation. The BiMP results not only detected chromosomal regions that became hypo- and hypermethylated in met1-3, but also confirmed at the entire genome level that de novo methylation in met1-3 is biased toward methylation of CHH sequences. This was formerly ascertained at only a few selected loci (Mathieu et al. 2007). Further, the BiMP data allowed us to select several hypermethylated loci and subsequently detect that the aberrant de novo methylation in met1-3 can occur at CG dinucleotides that were originally not methylated in the wild type, while regions that lost CG methylation indeed retain a hypomethylated status. This has not been reported before.
The next step is to apply BiMP technology to other organisms. For larger genomes, the technology may need adaptations for taking into account the increased repetitiveness and/or the increased mCG levels, as in mammals. However, given the success with small amounts of Arabidopsis material, characterization of methylation patterns in small tissue samples or during embryonic development, where material amounts are limited, could be approached. Finally, it is easy to envisage applying the “fourN” amplification method of bisulfite-converted DNA for ultra-high-throughput sequencing to allow genomic DNA methylation detection at single-base resolution. Although this resolution is the ultimate goal, ultra-high-throughput sequencing methods are not selective, are expensive, and require considerable hardware and software support. Hence, the approach demonstrated here may remain a viable option for analyzing many biological samples where DNA methylation needs to be examined. Obviously, a limitation of the experiments described here is the use of standard arrays. Future DNA methylation analyses, especially for a chosen set of diagnostic targets, could be performed using pyrosequencing or custom arrays with methylation-specific oligonucleotides. This should add enormously to the sensitivity and specificity of BiMP.
Methods
Plant material and DNA extraction
Plant growth conditions were as previously described (Mathieu et al. 2007). DNA from leaf material combined from nine individuals per sample was CTAB extracted, isopropanol precipitated, and RNase A and Proteinase K treated, as previously described (Warnecke et al. 2002). After enzymatic treatment, the DNA was purified by phenol/chloroform/isoamyl alcohol (25/24/1, Sigma) extraction and ethanol precipitation. DNA quantity was estimated using a TKO100 fluorometer (Hoefer Scientific) with Hoechst dye 33528 (Polysciences). Its purity was measured in a Genequant pro spectrophotometer (Amersham Pharmacia), and its integrity was assessed electrophoretically.
Bisulfite conversion
DNA aliquots of 4 μg were digested to completion with the DraI endonuclease (Promega) followed by extraction with phenol/chloroform/isoamyl alcohol, and ethanol precipitation. After two 70% ethanol washes, the pellet was resuspended in 23 μL of water, and 1-μL aliquots were again analyzed for concentration, digestion, and purity as described above. The sample concentrations were adjusted with water to 100 ng/μL. Aliquots of 20 μL (2 μg) of each DNA sample were subjected to bisulfite treatment using the EpiTect Bisulfite Modification Kit (Qiagen), which includes a DNA protective buffer. The degree of conversion was determined by sequencing a region of the PHAVOLUTA locus that lacks mCs in the wild type (Bao et al. 2004). Only samples with conversion levels >98% were used for random genomic amplification (data not shown).
DNA amplification
For random genomic amplification, one-twentieth of the bisulfite-converted samples (corresponding to maximally 100 ng) were ethanol precipitated, resuspended in 7 μL of water, and subjected to random primer extension. To each sample, 2 μL of 5× Sequenase buffer (USB) and 1 μL of primer “sixN” (40 μM) (GTTTCCCAGTCACGATCNNNNNN) or primer “fourN” (40 μM) (GTTTCCCAGTCACGATCNNNN), for the “sixN” or “fourN” methods, respectively, were added. In addition to the bisulfite-converted DNA samples, controls of genomic DNA prepared in parallel, but without bisulfite conversion, and template-free controls (“no-template control”) were prepared. The samples were placed in a thermal cycler, denatured for 2 min at 94°C and cooled to 10°C. During the 10°C hold (lasting 5 min), 5 μL of the elongation reaction mix was added to each sample and samples were mixed gently. The elongation reaction mix for one sample consisted of 1 μL of 5× Sequenase buffer (USB), 1.5 μL of a dNTP mixture (10 mM each of dATP, dCTP, dGTP, dTTP), 0.75 μL DTT (0.1 M), 1.5 μL of bovine serum albumin (500 μg/mL), and 0.3 μL of Sequenase (13 U/μL) (the elongation reaction mix was prepared on ice). To anneal random primers, the temperature was slowly increased (0.05°C sec−1) to 37°C and extension was allowed to occur for 8 min. The denaturation, Sequenase addition (0.9 μL of Sequenase and 0.3 μL of Sequenase dilution buffer), primer annealing, and elongation steps were repeated per sample. After the elongation cycles, sample volumes were adjusted to 60 μL. Half of the material (30 μL) was subjected to PCR amplification with primers complementary to known sequences introduced during the random priming reaction (GTTTCCCAGT CACGATC). For each sample, PCR amplification was performed in three separate aliquots (due to volume limits in PCR reactions). A reaction mix contained 10 μL of elongation reaction, 8 μL of 25 mM MgCl2, 20 μL of 5× PCR amplification buffer, 5 μL of a dNTP mixture (10 mM each of dATP, dCTP, dGTP, 8 mM dTTP, and 2 mM dUTP), 1 μL of primer (100 pmol/μL), 1 μL of GoTaq DNA polymerase (Promega 10 U/μL), adjusted to 100 μL with water. Samples were denatured for 3 min at 94°C followed by 30 cycles of 30 sec at 94°C; 30 sec at 40°C; 30 sec at 50°C; 1 min at 72°C followed by 10 min at 72°C. Each reaction was purified using the GenElute PCR clean-up kit (Sigma), and the three replicate reactions per sample (100 μL of eluate) were combined (300 μL final volume). DNA properties were assessed as described above, except that 3-μL aliquots were loaded for gel electrophoresis.
Post-amplification quality assessment
The randomly amplified reaction product was assessed for amplification bias using locus-specific PCR amplification and a dot blot hybridization approach (see Fig. 1). Briefly, for each sample, 1 μL of the final reaction product was subjected to a nested, locus-specific PCR amplification assay, as previously described (Mathieu et al. 2007). Primer sequences are listed in Supplemental Table 4. The PCR reaction products (10 μL per lane) were visualized in a 1% agarose gel stained with ethidium bromide.
To further evaluate the genome representation in randomly amplified reaction products, dot blot hybridizations were performed. The macroarray was prepared using a MINIFOLD 1 SRC 96-D vacuum filtration system (Schleicher & Schuell) with Hybond-N nylon membranes (Amersham Pharmacia) following the manufacturer’s protocol. Target probes (membrane-bound) were generated by spotting 200 ng of purified, denatured PCR product. The bisulfite-converted multilocus target consisted of six replicate 180-bp repeat amplicons (a heavily methylated repetitive element) (Saze et al. 2003). The non-bisulfite-converted targets consisted of amplicons from the following repetitive elements: 180-bp repeat (Saze et al. 2003), repeat “C,” repeat “E,” and repeat “Z”. The “C” (CACTA-like transposase family), “E” (pseudogene), and “Z” (pseudogene) elements each represent unique sequences within the Arabidopsis genome (data not shown). Primer sequences are listed in Supplemental Table 4. The positive control target was 200 ng of FWA amplicons (Soppe et al. 2000). The query DNA samples (hybridized to the target probes) were prepared by labeling 20-ng aliquots of randomly amplified DNA samples, spiked with the positive control (0.1 ng of FWA non-treated Col amplicon), with [α-32P]dCTP, using random hexamer priming. Following overnight hybridizations, the membranes were washed and exposed as previously described (Mathieu et al. 2007).
DNA fragmentation, labeling, hybridization
Per hybridization, 9 μg of “fourN” amplified DNA was ethanol precipitated and resuspended in 39.5 μL of nuclease-free sterile water. Fragmentation and labeling were performed using the wild-type double-stranded DNA terminal labeling kit (Affymetrix P/N 900,812). Per sample, 4.8 μL of 10× cDNA fragmentation buffer, 1.5 μL of UDG (10 U/μL), and 2.25 μL of APE 1 (100 U/μL) were added and briefly centrifuged. The fragmentation reaction was incubated for 2 h at 37°C, denatured for 2 min at 93°C and cooled for at least 2 min to 4°C. The samples were mixed, briefly centrifuged, and 45 μL was transferred to a new, sterile PCR reaction tube. Evaluation of the fragmentation reaction was performed with 1-μL aliquots of the remnant sample using a Bioanalyzer 2100 with the RNA 600 LabChip kit (Agilent) (see Supplemental Fig. 1D), as directed by the manufacturer.
A DNA-labeling reaction contained 12 μL of 5× terminal deoxynucleotidyl transferase (TdT) buffer, 2 μL of terminal deoxynucleotidyl transferase, 1 μL of DNA-labeling reagent (5 mM), and the DNA sample (total volume 60 μL). Labeling was performed for 60 min at 37°C, terminated by heating for 10 min to 70°C, and cooled for at least 2 min to 4°C. To the 60 μL of fragmented and labeled DNA, 4 μL of 3 nM B2 control oligonucleotides (Affymetrix), 12.5 μL of 20× RNA hybridization spike controls, 120 μL of 2× hybridization buffer, 16.8 μL of DMSO, and 36.7 μL of water were added and samples were heat denatured for 5 min at 99°C, cooled for 5 min to 45°C, and centrifuged at 13,000 rpm for 5 min. Each hybridization was performed by transferring 200 μL of sample to a pre-hybridized GeneChip Arabidopsis Tiling 1.0R array (Affymetrix) for 16 h at 45°C, as recommended by the manufacturer.
Data processing and analysis
Hybridization data, in the CEL file format, were analyzed using the Tiling Analysis Software (TAS version 1.1.02, Affymetrix). A probe analysis was performed using the perfect match/mismatch intensities. The dataset referred to in the Results section and figures as “non-treated,” “BiMP ColBS+,” and “BiMP met1-3BS+” were obtained using the one-sample detection analysis on the three biological replicate CEL files. The data were quantile normalized and scaled to a median intensity of 100. The log2-transformed estimated signal intensities were saved as BAR files. The correlation coefficients between the three replicate datasets per entry (Fig. 2) and the histogram distributions (Supplemental Fig. 2) were also obtained with TAS, as described in the user manual (Affymetrix). The chromosome-wide average signal intensities (Fig. 3A; Supplemental Fig. 3) were computed and plotted in the R statistical environment (http://www.r-project.org/) from a sliding-window algorithm (window length of 100 kb, slid by 10 kb).
The novel DNA methylation polymorphism analysis was initiated by performing a two-sample detection analysis in TAS. For the “BiMP ColBS+ normalized” and “BiMP met1-3BS+ normalized” datasets, the respective bisulfite-converted datasets (treatment group) were compared against the nontreated reference dataset (control group). The data were quantile normalized together using the parameters described above. The DNA methylation polymorphism analysis was completed within the Integrated Genome Browser (IGB version 3.62, Affymetrix). The normalized Col and met1-3 bisulfite-converted signal intensity files were imported into IGB to create difference graphs between the signal intensities. The hypomethylation graph presents the decrease in intensity of met1-3BS+ relative to ColBS+; the hypermethylation graph presents the increase in intensity of met1-3BS+ relative to ColBS+. For each difference graph, hypomethylated and hypermethylated intervals, respectively, were detected using intensity values set to a threshold of 1 × 105 with a maximum gap of 80 and a minimum run to 40, resulting in a “sliding window” analysis at a 161-bp length. The offsets for thresholded regions started and ended at 12 and 13, respectively. Intervals within the difference graphs >4.0 were reported as “hypomethylated” and “hypermethylated,” based on the analysis at known methylation polymorphisms (see Results) and sequence motifs per interval were calculated within the R statistical environment (http://www.r-project.org/; scripts available upon request). Processing and analysis of the mCIP data files for the methylation-enriched hybridizations of the wild type and met1-3 (http://www.ncbi.nlm.nih.gov/projects/geo/, accession no. GSE5094) were analyzed in TAS using the previously described parameters (Zhang et al. 2006). Within IGB, all signal intensity graphs were displayed in the range 0–11 (log2 scale).
Validation of novel DNA methylation polymorphisms
The bisulfite sequencing experiments (Fig. 5; Supplemental Figs. 5, 12) were performed with the same bisulfite DNA preparations used for the hybridizations and PCR-based cloning on five to 11 independent clones, and analysis was performed as previously described (Mathieu et al. 2007). The primers are listed in Supplemental Table 4. The methylation levels per sequence motif (CG, CNG, and asymmetric CHH), referred to in the figures as “% methylated,” were calculated by dividing the number of nonconverted cytosines by the total number of cytosine positions within the assay.
Acknowledgments
We thank Dr. Olivier Mathieu for review of the manuscript. This work was supported by grants from the Swiss National Science Foundation (3100A0-102107), the European Commission through The Epigenome (LSHG-CT-2004-503433), and TAGIP (018785).
Footnotes
-
↵3 Corresponding author.
↵3 E-mail jon.reinders{at}bioveg.unige.ch; fax 41-22-379-3107.
-
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to the Gene Expression Omnibus (GEO) at NCBI under accession no. GSE9051.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.7073008
-
- Received August 24, 2007.
- Accepted November 26, 2007.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








