
Punctuated duplication seeding events during the evolution of human chromosome 2p11
- Julie E. Horvath1,5,
- Cassandra L. Gulden1,
- Rhea U. Vallente1,4,
- Marla Y. Eichler1,
- Mario Ventura2,
- John D. McPherson3,
- Tina A. Graves3,
- Richard K. Wilson3,
- Stuart Schwartz1,
- Mariano Rocchi2, and
- Evan E. Eichler1,6,7
- 1 Department of Genetics and Center for Human Genetics, Case Western Reserve University School of Medicine and University Hospitals of Cleveland, Cleveland, Ohio 44106, USA
- 2 Sezione di Genetica, DAPEG, University of Bari, 70126 Bari, Italy
- 3 Washington University School of Medicine Genome Sequencing Center, St. Louis, Missouri 63108, USA
- 4 Washington State University School of Molecular Biosciences, Pullman, Washington 99164, USA
- 5 Institute for Genome Sciences and Policy, Duke University, Durham, North Carolina 27708, USA
- 6 Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA
Abstract
Primate genomic sequence comparisons are becoming increasingly useful for elucidating the evolutionary history and organization of our own genome. Such studies are particularly informative within human pericentromeric regions—areas of particularly rapid change in genomic structure. Here, we present a systematic analysis of the evolutionary history of one ∼700-kb region of 2p11, including the first autosomal transition from pericentromeric sequence to higher-order α-satellite DNA. We show that this region is composed of segmental duplications corresponding to 14 ancestral segments ranging in size from 4 kb to ∼115 kb. These duplicons show 94%–98.5% sequence identity to their ancestral loci. Comparative FISH and phylogenetic analysis indicate that these duplicons are differentially distributed in human, chimpanzee, and gorilla genomes, whereas baboon has a single putative ancestral locus for all but one of the duplications. Our analysis supports a model where duplicative transposition events occurred during a narrow window of evolution after the separation of the human/ape lineage from the Old World monkeys (10–20 million years ago). Although dramatic secondary dispersal events occurred during the radiation of the human, chimpanzee, and gorilla lineages, duplicative transposition seeding events of new material to this particular pericentromeric region abruptly ceased after this time period. The multiplicity of initial duplicative transpositions prior to the separation of humans and great-apes suggests a punctuated model for the formation of highly duplicated pericentromeric regions within the human genome. The data further indicate that factors other than sequence are important determinants for such bursts of duplicative transposition from the euchromatin to pericentromeric regions.
Human pericentromeric and subtelomeric regions, much like the majority of the Y chromosome, have long been viewed by many as “genetic wastelands” (Skaletsky et al. 2003) due to the fact that they are composed of large complex blocks of heterochromatic sequences and contain few genes (Donze and Kamakaka 2002). Recent studies suggest that understanding these transition regions will provide us a more complete picture of human genome architecture and the relationship of chromosome structure and function (She et al. 2004a). Despite recent advances in genome sequencing and the finishing of human euchromatin (International Human Genome Sequencing Consortium [IHGSC] 2004), the structure of these regions remains largely incomplete (Eichler et al. 2004). Sequence gaps are particularly enriched within pericentromeric regions, and most chromosome sequences fall short of bridging classically defined (Manuelidis 1978; Willard and Waye 1987; Willard 1991) heterochromatic sequences and euchromatin.
More recently, a handful of laboratories have extended efforts to include heterochromatic transition regions (Bailey et al. 2001; IHGSC 2001; Schueler et al. 2001; Rudd and Willard 2004; She et al. 2004a). From these and other efforts, we now understand that more than half of all human chromosomes contain segmentally duplicated sequences, primarily found in pericentromeric or subtelomeric regions. A noticeable reduction in transcription is observed within the most proximal 1 Mb portion of the duplication region, suggesting that some heterochromatic properties extend beyond α-satellite DNA. These duplications range in size from 1 kb to more than half a megabase and typically originate from euchromatic regions of the genome (She et al. 2004a). A few pericentromeric duplications have been characterized in detail, although the mechanism for their dispersal is still largely unknown (Guy et al. 2000, 2003; Ji et al. 2000; Bailey et al. 2001; Horvath et al. 2001; Samonte and Eichler 2002). A highly nonrandom distribution of duplications within pericentromeric regions has been noted with both quiescent and active regions of duplication for specific human chromosomes (She et al. 2004a).
Limited comparisons of pericentromeric regions among closely related primates suggest extraordinary dynamism where duplication, deletion, and rearrangement of large segments of DNA occur at an unprecedented scale (Eichler et al. 1996, 1997; Regnier et al. 1997; Zimonjic et al. 1997; Orti et al. 1998; Horvath et al. 2000b, 2003; Crosier et al. 2002). These findings have suggested that the actual number of “chromosomal rearrangements” among primates far exceed expectations based on the comparison of primate karyotypes. Limited phylogenetic analyses of a small number of segmental duplications (Eichler et al. 1997; Orti et al. 1998; Horvath et al. 2000b; Luijten et al. 2000) support a two-step model for their origin whereby initial rounds duplicate portions of the euchromatin to a specific pericentromeric “acceptor region.” Subsequent duplication events move larger blocks of duplication (often made of several blocks of initial duplication) among the acceptor regions.

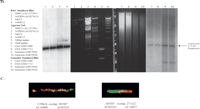
2p11 Duplicon architecture. (A) A schematic representation of the duplicon architecture (colored bars) is shown in reference to an ideogram of chromosome 2 and ∼700-kb BAC minimal tiling path. The black bar represents α-satellite sequence (∼175 kb), while light gray bars denote various pericentromeric-specific interspersed repeats (PIRs). Other enriched pericentromeric repeat sequences are indicated: C=CAAAAAG repeat, G=CAGGG, R=REP522, and T=TAR1 repeats (Smit 1996). Below the BAC tiling path are results of database searches using this entire sequence (represented by NT_034508) against the human genome (build34, July 2003). All pairwise alignments (>5 kb and >90%) to this segment are shown to other regions of the genome as indicated by the chromosome number and approximate position in megabases (ancestral loci are denoted by cytogenetic band position). A color scheme encodes the average percentage sequence identity for each alignment block (red, 99%; orange, 98–99%; yellow, 97–98%; green, 96–97%; blue, 95–96%; indigo, 94–95%; and violet, 90–94%). (B) Sequence overlaps were confirmed by Southern analysis between BAC clone and genomic DNA. An example of validation is shown for overlap D (between AC127391 [R11–389I13] and AC027612 [R11–165D20]). A PCR-generated probe (165D20–6n7) (Supplemental Table 2) was hybridized. The expected 2.2-kb band is observed in multiple overlapping BACs (389I13, 165D20, 34O12, and 1430E12) in addition to the chromosome 2 hybrid and genomic DNA samples. Note: An additional lower band is observed in the genomic DNA samples compared with the monochromosomal hybrid DNA samples, indicating that at least one additional copy of the GGT1 duplicon exists within the human genome. (C) Extended fiber FISH validating overlap (in yellow) of the three most proximal BACs in a chromosome 2 hybrid cell line (GM11712). Results in a second chromosome 2 hybrid line (GM11686) and total human cell lines showed similar results (data not shown).
In an effort to provide insight into these complex regions of our genome, we conducted a detailed molecular evolutionary analysis of a 700-kb pericentromeric region of human chromosome 2p11. This human chromosome is particularly remarkable since it contains a large number of highly identical inter- and intrachromosomal segmental duplications. It is also noteworthy as the only chromosome to have emerged in the human lineage as a result of a chromosome fusion (Ijdo et al. 1991; Fan et al. 2002). There were two main objectives of this research: (1) to characterize the organization of the 2p11 pericentromeric region up to and including higher-order α-satellite repeats and (2) to assess the evolutionary origin and the timing of the duplication events in primate evolution. Our previous pilot analysis of 2p11 indicated that this type of organization was a property common to many pericentromeric regions. Therefore, 2p11 provides a model for the organization of many human pericentromeric regions containing interchromosomal duplications, and gives us insight into the general mechanism for their formation.
Results
Sequence, assembly, validation, and annotation of the 2p11 pericentromeric region
We constructed a physical map and sequenced 700 kb of the most proximal portion of the short arm of human chromosome 2. The presence of high-identity duplications to multiple regions of the human genome complicates sequence and assembly of these regions (She et al. 2004b). The organization and representation of human chromosome 2 was, therefore, validated by several independent methods, including analysis of sequence overlaps (see tiling path in Fig. 1A), genomic Southern blot analysis (Fig. 1B), two-color FISH experiments (Fig. 1C), and paralogous sequence tagging of monochromosomal DNA (Supplemental Table 1; Supplemental Methods). The assembled sequence included some of the largest (175 kb) contiguous transition sequence into human α-satellite DNA. Several lines of evidence indicate that we have successfully traversed higher-order sequences from chromosome 2 (Supplemental Methods; Supplemental Fig. 1A,B).
We annotated the duplication content by using a variety of computational methods. Seven regions with conserved exon/intron structure were identified within the 2p11 sequence although none contained a complete complement of exons as predicted by the full-length transcript. In each case, the full-length gene mapped to another region of the genome. These were termed duplicons (segmental duplications where the ancestral origin can be determined). Since this search for ancestral duplicons was not limited to sequences outside of defined pericentromeric regions (5 Mb around the centromere), we identified two additional duplicons (GGT1 and IGSF3) that were not identified previously (She et al. 2004a). The 2p11 duplicons included CHK2 (checkpoint kinase 2) from 22q12, an unknown gene from 4q24, ALD (adrenoleukodystrophy) from Xq28, GGT (γ-glutamyltransferase 1) from 22q11, IGSF3 (immunoglobulin superfamily 3) from 1p13, MLL3 (myeloid/lymphoid leukemia 3) from 7q36, and LSP1 (lymphocyte-specific protein 1) from 11p15. With the exception of LSP1, none of these segments showed any evidence of transcription based on sequence similarity searches of human EST databases.
To identify the putative boundaries of each duplication, we examined all underlying pairwise alignments for the entire region by using PARASIGHT (http://humanparalogy.gs.washington.edu/parasight). This allowed us to obtain the minimally shared segment for each region (Bailey et al. 2002) and facilitated the identification of seven more putative duplicons (PIR4, 11q14, 12p11, λ immunoglobulin (Igλ), 10q26, 4p16.1, and 4p16.3) (Fig. 1A; Table 1) within 2p11. Five of these were previously identified by mouse synteny mapping, but two (PIR4 and Igλ) were excluded due to their location within a pericentromeric region (She et al. 2004a). All 14 of the identified duplicons represent duplicated segments from seven different human chromosomes, exhibit 94%–98.5% identity to the putative ancestral loci, and range in size from <4 kb to >115 kb. Three of these duplicons (IGSF3, GGT1, and LSP1) were shown previously to exist on chromosome 2 by FISH, but detailed analyses into their genomic organization or evolutionary histories were lacking (May et al. 1993; Tassone et al. 1995; Saupe et al. 1998; Ruault et al. 1999).
2p11 Duplicon sequence properties
Previous studies have suggested that GC-rich and Alu repeat elements are enriched at the boundaries of duplication (Eichler et al. 1999; Horvath et al. 2000a; Chen and Li 2001) and implicated these as playing a role in the process of segmental duplication (Bailey et al. 2003). In this study we were able to distinguish both donor and acceptor loci (phylogenetically and by comparative FISH). Based on sequence comparison to the ancestral locus, we were able to define 38 donor and acceptor boundaries. Analysis of duplicon termini in 2p11 (Fig. 1A) indicates that GC-rich repeat sequences (CAGGG, CAAAAG, TAR, and REP522) (Smit 1996) occur within 1 kb for at least five of 19 of the acceptor regions. No enrichment of these elements was noted in the vicinity of the donor regions. If we narrowed the junctions to a 5-bp window (Table 2), we found that 15 of 38 (39%) of the donor boundaries and 16 of 38 (42%) acceptor regions show the presence of an Alu S or Y repeat sequence at the junction. This Alu enrichment is consistent with previous reports and suggests that Alu repeats have played an important role in initializing pericentromeric seeding events while GC-rich elements contribute to the pericentromeric swapping. At present, there is, however, only indirect evidence for such associations.
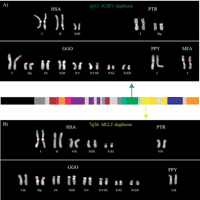
Comparative primate FISH of individual duplicons. Two examples of comparative metaphase FISH experiments for the (A) IGSF3 (dark green) duplicon from 1p13 and the (B) MLL3 duplicon (in yellow) from 7q36 are shown. Extracted metaphases for five primates are shown after hybridization with probes corresponding to the two duplicons: HSA indicates H. sapiens; PTR, P. troglodytes; GGO, G. gorilla; PPY, P. pygmaeus; and MFA, M. fascicularis. Both sets of experiments show multiple signals among humans and the great-apes with a single signal in the Old World monkey macaque. These results are consistent with the phylogenetic and comparative genomic hybridization experiments that suggest a duplication of the ancestral locus <23 Mya. All chromosomal designations are with respect to the human phylogenetic group (McConkey 2004).
Evolutionary analysis of 2p11 duplications
A three-pronged approach was used to reconstruct the evolutionary history of this region. Each of the 14 duplicons (defined above) was treated independently in this analysis. Comparative FISH was used to delineate the origin, dispersal, and copy number variation among closely related primate species. Screening of genomic libraries from nonhuman primates was used as a mapping approach to refine ancestral locations of each duplicon based on comparison of the clone ends to the human genome sequence (see below). Phylogenetic analysis of sequence from each duplication was then used to reconstruct the likely order and timing of the individual duplications during the past 25 million years (Myr) of human genome evolution.
We performed comparative FISH against metaphase chromosomes of four hominoid species (Homo sapiens, Pan troglodytes, Gorilla gorilla, and Pongo pygmaeus) and one Old World monkey representative (Papio hamadryas or Macaca fascicularis). Genomic probes were prepared for all duplicons >15 kb in size, and hybridization results are summarized in Table 3 (for a representative set of experiments, see Fig. 2). In general, our FISH results indicate a reduction in copy number as probes are hybridized to orangutan and baboon. Interestingly, in several cases, no signals were observed among baboon or macaque. Although not all probes are single copy in orangutan, these results verify many of the putative duplicon ancestral positions as predicted by the origin of the expressed gene (see results for 4q24, Xq28, IGSF3, and MLL3 in Table 3). Reciprocal experiments were conducted with baboon BACs representing each duplicon on baboon and human metaphase chromosomal spreads. Duplicons 11q, 12p, 4q24, ALD (from Xq28), and IGSF3 (from 1p13) were verified to be ancestral loci based on the observation of a single signal in baboon (data not shown).
Comparative FISH results
Since FISH experiments did not always yield a reliable signal in orangutan or baboon, we conducted genomic library hybridizations as a secondary means to refine the ancestral origin more precisely. A PCR probe (for location, see Fig. 1A; for sequence, see Supplemental Table 2) was designed within each duplicon and was used to screen large-insert genomic BAC libraries from orangutan (CHORI-253) and baboon (RPCI-41). Based on the genomic coverage and the number of positively hybridizing BACs, we estimated the copy number for each duplicon within each primate species (Table 4; Supplemental Methods). With the exception of the Igλ segment (which maps to a tandem gene cluster), the PIR4 segment (which was not identified in the baboon), and the LSP1 duplicon (which apparently has undergone an independent duplication expansion), 11 out of the 14 duplicons mapped to a single locus in either orangutan or baboon (Table 4). Orangutan and baboon BACs corresponding to each single site were end-sequenced, and the sequences were aligned to the human genome reference sequence by using BLAST (build 34, NCBI, July 2003) (Supplemental Tables 3, 4). With the exception of orangutan IGSF3 BACs, primate BAC end-sequences from each duplicon corresponded to human sequence located at the putative ancestral location.
Summary of BAC hybridization results
To provide a more precise estimate of duplication timing, we performed a phylogenetic analysis based on primate comparative sequencing of each duplicon as described previously (Horvath et al. 2003). By utilizing PCR assays designed to noncoding 2p11 human reference sequence, orangutan and baboon BACs were PCR amplified, and the products were directly sequenced with multiple primer pairs within each duplicon. We constructed a neighbor-joining phylogenetic tree for 11 of the duplicons where complete sequence information could be obtained (Fig. 3). Genetic distances are indicated in Table 5 and were used to calculate the ancestral nucleotide substitution rate specifically for each duplicon. These substitution rates range between 1.13 and 1.83 × 10–9 substitutions/site/yr and are generally consistent with estimates from other duplicated segments (Eichler et al. 1999; Liu et al. 2003). These values were then used to calculate seed and swap times corresponding to initial duplication of each donor segment and subsequent dispersal of these segments to other pericentromeric regions.
Genetic distances summary
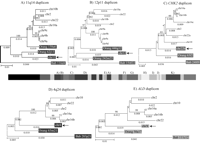
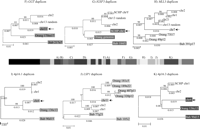
Phylogenetic trees for 2p11 duplicons. A neighbor-joining tree was constructed for each individual duplicon as shown above and below the gray schematic of the 2p11 duplicons (A–K). See Figure 1 for corresponding colored boxes. Gray boxes outline ancestral human, orangutan (Orang), and baboon (Bab) sequence taxa within the phylogenetic trees. Ancestral human sequences are also marked with an arrow. Branch lengths are proportional to the number of nucleotide changes between taxa and are indicated below each respective branch. An asterisk next to or below a branch length indicates a branch length of 0.001. Bootstrap values >90 from 1000 replicates are indicated above each corresponding branch. Sequence data from baboon and orangutan outgroups were obtained from large-insert BAC clones (CHORI-253 and RPCI-41) or total genomic DNA.
Ten of the 11 tree topologies are consistent with a major duplication seeding event occurring after the separation of Old World monkey and great-ape lineages (<23 million years ago [Mya]). All 10 phylogenies clearly distinguish two major events: an ancestral event (termed an ancestral duplicative transposition) followed by a series of secondary duplications (pericentromeric swapping) that group all human paralogs. Bootstrap support distinguishing these events ranges from 96–100 (Fig. 3). The LSP1 duplicon is the only locus that is inconsistent with this model of evolution. In some cases, we observed similarities in the tree topology based on spatial proximity of the ancestral duplicons within 2p11. The first three duplications (for PIR4 tree, see Horvath et al. 2003) nearest the human centromere, for example, show evidence of duplication of the ancestral locus prior to the divergence of the humans and the great-apes from the Old World monkey lineage as evidenced by progenitor duplicates in the orangutan lineage. In general, evolutionary genetic distance estimates between human ancestral and paralogous loci (0.03–0.06) are significantly less than the genetic distance between the human ancestral locus and the corresponding baboon locus (0.05–0.08) (Fig. 4; Table 5). By using locus-specific substitution rates, we calculated that the initial duplication of the ancestral locus occurred between 9 and 19 Mya. Although secondary dispersal events occurred ∼3–11 Mya (Fig. 4; Table 5), there is no evidence of a novel ancestral duplicative transposition event having occurred over the past 9 Myr within this region of 2p11.
Discussion
We present one of the most comprehensive evolutionary analyses, to date, of a human centromeric transition region. We have extended the model of pericentromeric duplication by systematically tracking the origin and timing of a series of duplicons located within a 700-kb pericentromeric region of 2p11 (She et al. 2004a). Our goal was to reconstruct the evolutionary history of this region by using a combination of phylogenetic, genomic, and comparative FISH approaches. Our study provides compelling evidence for an evolutionarily punctuated movement of duplicated material 10–20 Mya for the majority of the 2p11 pericentromeric region. Although we can not preclude the existence of more ancient duplications of euchromatin that have been deleted/diverged before this time period, the identification of more recent ancestral duplicative transpositions should have been trivial to detect. None, however, were identified within this portion of 2p11.
Previous analyses have suggested that pericentromeric regions have been formed via the duplication of euchromatic segments that have colonized pericentromeric DNA over the past 30 Myr of evolution (Eichler et al. 1996; Jackson et al. 1999; Horvath et al. 2000b, 2003; Bailey et al. 2002; Crosier et al. 2002; She et al. 2004a; Locke et al. 2005). This duplicative transposition of euchromatic segments into pericentromeric regions (which we have termed “pericentromeric seeding”) has led to the formation of complex mosaics of segmental duplications consisting of juxtaposed duplicons from diverse euchromatic positions. Secondary duplications of larger mosaic blocks (termed “pericentromeric swapping” events) occurred subsequently, leading to differential distribution of these blocks among the great-ape and human pericentromeric regions. Detailed analyses of pericentromeric regions (10p11, 10q11, 15q11, 2p11, and 16p11), as well as more global computational analysis, suggest that this is a general principle of human genome evolution (Jackson et al. 1999; Guy et al. 2000, 2003; Horvath et al. 2000b; Locke et al. 2005). Our extended analysis of 2p11 confirms this two-step model (Fig. 5) but also indicates that most euchromatic seeding events occurred over a more narrow window of evolutionary time than previously appreciated (Guy et al. 2000, 2003; Bailey et al. 2001, 2002).
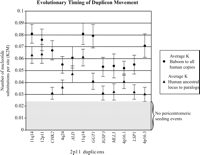
Sequence divergence of 2p11 duplicons. The graph compares the average divergence (substitutions per site, Kimura two-parameter model with standard error measurements) for baboon and all human duplicate copies (circles) to the average divergence for the human ancestral locus to all human pericentromeric copies (triangles). The former provides a locus-specific estimate of the effective number of substitutions since the divergence of Old World monkeys and human lineages (∼23 Mya), while the latter provides an estimate of the timing of the initial duplication event. With the exception of LSP1, the baboon copy corresponds to a single (nonduplicated) locus. The data are consistent with an initial duplicative transposition of the ancestral locus for all loci after separation of the Old World and human lineages. No duplications from an ancestral locus are observed within this 700-kb region which show <0.03 substitutions/per site. This suggests a cessation of euchromatic colonization of this region ∼10 Mya.
Results from comparative FISH of 2p11 duplicons indicate that many segments were originally duplicated after the divergence of the human and baboon lineages (∼23 Mya), but before the divergence of human and the African great-apes (∼8 Mya) (Fig. 2; Table 3). The phylogenetic data agree closely with the comparative FISH data. The genetic distance, for example, between human and baboon sequence ranges from 0.052–0.081, while the evolutionary distance between the human euchromatic ancestral locus and pericentromeric paralogs ranges from 0.03–0.064 (Fig. 4; Table 5). Based on relative rate tests and individual calibration for the substitution rate of each locus, these distances translate into pericentromeric seeding events that occurred 10–20 Mya. As expected, our genomic studies occasionally identified duplicated sequence among the orangutan great-apes (thought to have diverged 12–14 Mya) (Fig. 3). No additional evidence of euchromatic to pericentromeric seeding events could be identified within human 2p11 after the separation of humans from chimps and gorillas, although secondary duplication events (pericentromeric swapping) are readily observed.
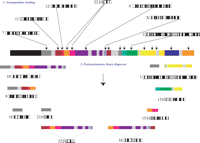
A model for the acquisition and dispersal of 2p11 duplicons. An expanded two-step model is shown to explain the current organization of 2p11. First, a burst of DNA duplicative transposition events occurs in the common ancestor of humans and apes (10–20 Myr), creating a large mosaic region consisting of at least 14 duplicons. During the radiation of humans and African great-apes (4–8 Mya), a series of secondary duplications disperse larger cassettes to other pericentromeric regions, leading to quantitative and qualitative differences of each larger block within different lineages. More recent transposition events suddenly cease or are no longer fixed during this second phase.
It is unclear why pericentromeric seeding events occurred so frequently during this period of human/great-ape evolutionary history. It is also unclear why they suddenly cease, at least in the case of 2p11. One possible scenario may be that certain regions of the genome are permissive to segmental duplication events only at specific periods of time. The permissive nature may relate to evolutionary changes in transcriptional activity or the chromatin configuration of these regions. In such a scenario, one might expect to find pericentromeric regions with younger or older duplicons depending on differences in the chromatin context in which they emerged. A global analysis of several pericentromeric regions confirms that, in general, younger (<8 Mya) pericentromeric seeding events are a relatively rare occurrence in the human genome (Bailey et al. 2002; She et al. 2004a; Locke et al. 2005). This is not to say that pericentromeric-to-pericentromeric duplications have not continued to occur more recently. Indeed, there are numerous examples of such pericentromeric swapping events that have emerged since the great-ape/human separation, and a few have been unambiguously shown to be lineage-specific events (Bailey et al. 2002). In addition, other nonpericentromeric regions of the human genome show ample evidence of more recent (<8 Mya) duplicative transposition events into acceptor regions (Johnson et al. 2001; Stankiewicz et al. 2004).
There are several other possible scenarios that may be put forward to explain this punctuated genome restructuring process. For example, it is interesting to note that the “shift” from pericentromeric seeding to pericentromeric swapping coincides with the emergence of higher-order α-satellite DNA (8 Mya) (Haaf and Willard 1998). This change in centromeric higher-order structure may have influenced ectopic recombination events among nonhomologous chromosomes, providing a mechanism for these secondary duplication events.
We cannot rule out the possibility that our view of the duplication process as “punctuated” is obscured by having an incomplete genome. If new seeding events are primarily restricted to the unsequenced p arms of acrocentric chromosomes, we may miss them entirely. There is a small amount of evidence that acrocentric p arms do harbor duplicons (Wohr et al. 1996; Eisenbarth et al. 1999; Hattori et al. 2000; Cserpan et al. 2002); however, their sequence identity attributes do not appear to differ significantly from what has been observed for other pericentromeric regions.
High-quality BAC-based sequence within pericentromeric regions has revealed a remarkable level of evolutionary dynamism. Comparative studies such as these provide valuable information into the evolutionary forces that have reshaped our genomes—forces that likely contribute to contemporary variation and disease. Detailed comparative sequencing of these regions, however, is required to address several of the hypotheses and models that we have put forward. While correct assembly of these regions is often a daunting task, we have demonstrated that such regions can be assembled and sequenced with available genomic resources (Horvath et al. 2000a). Unfortunately, the quicker method of sequence assembly, whole genome shotgun assembly, may preclude such rich evolutionary analyses as complex and duplicated regions will be incorrectly assembled or simply not represented (She et al. 2004b). Targeted comparative studies with large-insert clones from these regions promise to provide valuable insight into the evolution of our species and genome.
Methods
Computational analyses
Duplicon identification was conducted for each individual accession by using RepeatMasker (RepeatMasker version 07/13/2002; A. Smit and P. Green, http://ftp.genome.washington.edu/RM/RepeatMasker.html) sequence as query against the EST division of GenBank. All ESTs showing exon/intron structure to the query accession were used to identify UniGene clusters when available (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene). A representative EST from each UniGene cluster was used as query against nr (nonredundant) and htgs (high throughput genome sequence). All ESTs not belonging to a UniGene cluster were used as query individually. An accession with an identical match to the representative EST was considered the ancestral locus and was used to identify the chromosomal region in build34 for further comparisons of duplicon size and identity (Table 1). Optimal global alignments of BAC overlaps and ancestral loci to each 2p11 paralogous segment were generated by using the program ALIGN (Myers and Miller 1988). NT_034508 was used (in reverse orientation) for database searches of all paralogous and ancestral loci in build34. These hits were displayed by using PARASIGHT (http://humanparalogy.gs.washington.edu/parasight) (Fig. 1A).
PCR and sequencing
The BAC and cosmid clones used for PCR analysis were grown from single colony isolates in 5 mL overnight cultures. The DNA was isolated by using the Millipore (Millipore) or Perfectprep BAC 96 kit (Eppendorf) and resuspended in water. Approximately 15 ng BAC DNA (1/25 the total volume) and 15 ng of cosmid DNA (1/50 the total volume) were used in subsequent PCR assays. All PCR and sequencing conditions were previously described elsewhere (Horvath et al. 2003). BAC end sequencing reactions were conducted as previously described (She et al. 2004a). Cosmid end sequencing reactions were identical to BAC end reactions except that only 1/12 the total volume of cosmid DNA was used, and only 70 cycles of sequencing were conducted. We assessed the quality of all sequence data using PHRED/PHRAP/CONSED software (http://genome.wustl.edu).
Phylogenetic analysis
FASTA formatted sequences were obtained after comparison of both forward and reverse sequences from each PCR product using CONSED. All primate BAC sequences were searched against build34 to obtain all fully sequenced human copies. Sequence alignments were built by using CLUSTALW (version 1.82) (Higgins et al. 1996), and maximum parsimony, minimum evolution, and neighbor-joining methods were all used to construct phylogenetic trees by using MEGA (Molecular Evolutionary Genetic Analysis) v2.1 (http://www.megasoftware.net/) (Kumar et al. 2001). Although all three methods yielded trees with identical topology, neighbor-joining phylograms are shown because they allow for distance estimates between taxa. Neighbor-joining analysis was used with complete deletion parameters for all duplicon trees (Fig. 3) and pairwise deletion parameters for the α-satellite trees (Supplemental Fig. 1B) with 1000 bootstrap iterations. Tajima's relative rate tests (Tajima 1993) were used in MEGA (Kumar et al. 2001) to determine if rates of nucleotide substitution were constant between the three species (human, orangutan, and baboon). We estimated the number of substitutions/site/year (substitution rate) by correcting the divergence times for multiple substitutions using Kimura's two-parameter model (Kimura 1980). Divergence times of 23 Myr between the human and baboon lineages and 13 Myr between human and orangutan lineages were used. Duplication timing events were calculated by using the equation T=K/2r (Li 1997). The approximate seed time (in millions of years) was determined by multiplying the ancestral to paralog K value by 23 Myr (human to baboon divergence estimate) and dividing by the baboon to paralog K value. Swap times were calculated using the average K of all human paralogs in place of the ancestral to paralog K value.
Acknowledgments
We thank Lawrence Livermore National Labs and the UK HGMP Resource Centre for providing the cosmid library filters and clones. We thank Sean McGrath, Mandeep Sekhon, Andrew Grow, Jason Carter, and Laurie Christ for technical assistance and Dr. Norman Doggett for kindly providing access to chromosome 16 cosmid filters and clones. We thank Huntington F. Willard, Carol Stepien, Stuart Schwartz, Mitch Drumm, and Joe Nadeau for insightful discussions regarding all aspects of this work. We also thank Mary Schueler and Katie Rudd for helpful discussions regarding α-satellite DNA, and Lisa Chadwick for helpful suggestions with this manuscript. Chromosome ideograms for Figure 5 were obtained from the University of Washington Department of Pathology Web site: (http://www.pathology.washington.edu/research/cytopages/idiograms/human/). This work was supported, in part, by NIH grants HG002385 and GM58815 to E.E.E. In addition, we gratefully acknowledge Telethon, CEGBA (Centro di Eccellenza Geni in campo Biosanitario e Agroalimentare), MIUR (Ministero Italiano della Università e della Ricerca; Cluster C03, Prog. L.488/92), and the European Commission (INPRIMAT, QLRI-CT-2002-01325) for financial support. J.E.H. was supported in part by NIH GM08613, Genetics Training grant.
Footnotes
-
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. AY954301–AY954363.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3916405. Article published online before print in June 2005.
-
↵7 Corresponding author. E-mail eee{at}gs.washington.edu; fax (206) 685-7301.
-
- Accepted May 3, 2005.
- Received March 14, 2005.
- Cold Spring Harbor Laboratory Press








