
1274 Full-Open Reading Frames of Transcripts Expressed in the Developing Mouse Nervous System
- Maria F. Bonaldo1,
- Thomas B. Bair2,
- Todd E. Scheetz2,3,
- Einat Snir1,
- Ike Akabogu1,
- Jennifer L. Bair1,
- Brian Berger1,
- Keith Crouch1,
- Aja Davis1,
- Mari E. Eyestone1,
- Catherine Keppel1,
- Tamara A. Kucaba1,
- Mark Lebeck1,
- Jenny L. Lin4,
- Anna I.R. de Melo1,
- Joshua Rehmann1,
- Rebecca S. Reiter4,
- Kelly Schaefer1,
- Christina Smith1,
- Dylan Tack5,
- Kurtis Trout1,
- Val C. Sheffield1,6,
- Jim J-C. Lin4,
- Thomas L. Casavant2,3,5,7, and
- Marcelo B. Soares1,8,9,10,11
- 1 Department of Pediatrics, The University of Iowa, Iowa City, Iowa 52242, USA
- 2 Department of Center for Bioinformatics and Computational Biology, The University of Iowa, Iowa City, Iowa 52242, USA
- 3 Department of Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, Iowa 52242, USA
- 4 Department of Biological Sciences, The University of Iowa, Iowa City, Iowa 52242, USA
- 5 Department of Electrical and Computer Engineering, The University of Iowa, Iowa City, Iowa 52242, USA
- 6 Department of Howard Hughes Medical Institute, The University of Iowa, Iowa City, Iowa 52242, USA
- 7 Department of Biomedical Engineering, The University of Iowa, Iowa City, Iowa 52242, USA
- 8 Department of Biochemistry, The University of Iowa, Iowa City, Iowa 52242, USA
- 9 Department of Physiology and Biophysics, The University of Iowa, Iowa City, Iowa 52242, USA
- 10 Department of Orthopaedics, The University of Iowa, Iowa City, Iowa 52242, USA
Abstract
As part of the trans-National Institutes of Health (NIH) Mouse Brain Molecular Anatomy Project (BMAP), and in close coordination with the NIH Mammalian Gene Collection Program (MGC), we initiated a large-scale project to clone, identify, and sequence the complete open reading frame (ORF) of transcripts expressed in the developing mouse nervous system. Here we report the analysis of the ORF sequence of 1274 cDNAs, obtained from 47 full-length-enriched cDNA libraries, constructed by using a novel approach, herein described. cDNA libraries were derived from size-fractionated cytoplasmic mRNA isolated from brain and eye tissues obtained at several embryonic stages and postnatal days. Altogether, including the full-ORF MGC sequences derived from these libraries by the MGC sequencing team, NIH_BMAP full-ORF sequences correspond to ∼20% of all transcripts currently represented in mouse MGC. We show that NIH_BMAP clones comprise 68% of mouse MGC cDNAs ≥5 kb, and 54% of those ≥4 kb, as of March 15, 2004. Importantly, we identified transcripts, among the 1274 full-ORF sequences, that are exclusively or predominantly expressed in brain and eye tissues, many of which encode yet uncharacterized proteins.
The Brain Molecular Anatomy Project (BMAP) was initiated in 1998 by the National Institute of Mental Health (NIMH) and the National Institute of Neurological Disorders and Stroke (NINDS) as an interdisciplinary project to establish state-of-the-art technologies and informatics systems to decipher the molecular anatomy of the mammalian brain. One of the aims in the first phase of this project was the discovery of most transcripts expressed in the mouse brain, and the development of a comprehensive nonredundant arrayed collection of BMAP cDNAs and expressed sequence tags (ESTs). It was anticipated that such resources would greatly facilitate large-scale parallel analyses of gene expression studies aimed at localizing the site of expression of all BMAP transcripts in the brain. Toward this goal, we contributed ESTs that defined 28,000 of NCBI's mouse UniGene clusters, from ∼80,000 ESTs generated from a comprehensive collection of BMAP cDNAs representing 12 microdissected regions of the adult C57BL/6 mouse brain, spinal cord, and retina (T. Scheetz, M. Bonaldo, B. Berger, K. Crouch, N. Wu, J. Kasperski, M. Eyestone, J. Rehmann, C. Smith, T. Kucaba, et al., in prep).
In 2001, as part of a broader trans-National Institutes of Health (NIH) effort and in close coordination with the NIH-Mammalian Gene Collection (MGC) Program (Strausberg et al. 2002), we initiated the second phase of the BMAP with the objective of identifying and determining the complete and accurate protein coding sequence of a large number of transcripts expressed in the developing mouse nervous system. Forty-seven (NIH_BMAP) cDNA libraries were generated from size-fractionated cytoplasmic mRNA obtained from brain and eye tissue at multiple stages of embryonic development and at postnatal days 1, 5, and 15, using a novel approach that we developed for construction of full-length-enriched cDNA libraries. The 175,990 5′ ESTs, comprising 14,973 distinct clusters, were derived from these libraries based on alignment to the mouse genome. Of these, 7774 clones were tentatively identified as full-ORF-containing cDNAs, including 4223 transcripts novel to MGC. Further analysis of these 4223 clones resulted in the selection of 2084 cDNAs for full-insert sequence production, of which 1863 have been completed. Final analysis of these sequences led to the identification of 1274 NIH_BMAP full-ORFs, all of which have been submitted to National Center for Biotechnology Information (NCBI)/MGC.
Here we report the complete and accurate sequence of 1274 NIH_BMAP full-ORF-containing cDNAs. Thus, added to the 1019 full-ORF MGC sequences derived from NIH_BMAP cDNA libraries by the MGC sequencing team, NIH_BMAP full-ORF sequences correspond to ∼20% of all transcripts currently represented in mouse MGC (total of 10,295 nonredundant and 12,974 redundant mouse MGC sequences as of March 15, 2004; http://mgc.nci.nih.gov/). Most significantly, we show that NIH_BMAP clones constitute 68% of the mouse MGC clones ≥5 kb, and 54% of those ≥4 kb. In addition, we describe a new approach that we developed for construction of full-length-enriched cDNA libraries and successfully used for construction of 47 NIH_BMAP cDNA libraries. Furthermore, we present the results of an analysis that we performed with the sequences derived from the 1274 NIH_ BMAP cDNAs, to identify those in UniGene clusters with highest relative representations of ESTs derived from brain and eye tissues, respectively. This analysis reveals a number of transcripts that are predominantly expressed in the brain, and several others with distinctive expression in the eye, many of which encode uncharacterized proteins.
RESULTS AND DISCUSSION
A New Approach for Construction of Full-length-Enriched cDNA Libraries
A number of methods have been developed for construction of cDNA libraries enriched for full-length cDNAs, each presenting its own advantages and disadvantages (Maruyama and Sugano 1994; Suzuki et al. 1997; Carninci and Hayashizaki 1999; Carninci et al. 2000, 2001; Piao et al. 2001; Shibata et al. 2001; Suzuki and Sugano 2001, 2003). Enrichments achieved with these methods vary over a wide range, at least in part due to confounding factors pertaining to RNA integrity, nuclear RNA contamination, impeding RNA secondary structures, and characteristics and quality of critical components of the system, such as, but not limited to, the cloning vector, and the enzymes required for cDNA synthesis and cloning.
Despite all difficulties and inherent limitations, a great number of full-length-enriched human and mouse cDNA libraries have been produced, and Carninci's “CAP trapper” (Carninci and Hayashizaki 1999; Carninci et al. 2000, 2001, 2002; Shibata et al. 2001; Hirozane-Kishikawa et al. 2003), and Sugano's “Oligo-capping” (Maruyama and Sugano 1994; Suzuki et al. 1997; Suzuki and Sugano 2001, 2003) methods have proven invaluable. As a result, significant progress has been made toward the complete sequence characterization of both the human and the mouse transcriptomes (Okazaki et al. 2002; Strausberg et al. 2002; Carninci et al. 2003; Ota et al. 2004).
An important development in this arena was the establishment of the MGC Program, a trans-NIH initiative to generate a publicly available resource of accurately sequenced full-length ORF clones for all human, mouse, and rat genes (http://mgc.nci.nih.gov/). It is noteworthy that MGC's objective is not to obtain strictly full-length cDNAs, that is, complete copies of the mRNAs from the 5′ CAP to the 3′ poly(A) addition site, but rather full-ORF-containing cDNAs. At present, an important limitation of the MGC program is that it seeks to obtain only one representative full-ORF sequence from each transcription unit, despite the fact that multiple transcripts might be derived from any given transcription unit by virtue of utilization of more than one promoter, alternative splicing, and/or differential polyadenylation.
A significant development in deciphering the transcriptome was the creation of the Mouse BMAP, a trans-NIH initiative aimed at understanding gene expression and function in the nervous system (http://trans.nih.gov/bmap/index.htm). Among its objectives is the identification and sequencing of most transcripts expressed in the mouse brain. As part of this effort, and in coordination with the NIH-MGC Program, we began a project aimed at cloning, identifying, and determining the sequence of a large number of full-ORF-containing cDNAs, representing transcripts expressed in the developing mouse nervous system. This project provided us the opportunity to use and rigorously test an approach that we developed for construction of full-length-enriched cDNA libraries, which attempts to overcome a problem commonly observed in full-length cDNA libraries, that is, over-representation of full-length cDNAs derived from smaller transcripts and lack or disproportionate representation of full-length cDNAs derived from longer transcripts.
The approach we developed for construction of full-length-enriched cDNA libraries involves four principal steps: (1) size-fractionation and purification of high-quality cytoplasmic poly(A)+ mRNAs; (2) synthesis of oligo-dT-primed first-strand cDNA from each mRNA size-fraction, individually, using RNaseH- reverse transcriptase under optimized conditions to yield full-length cDNAs with short 5′ dT-tails; (3) size-selection and purification of double-stranded cDNAs according to the size range of the mRNAs in the size-fraction from which they originated; and (4) separate cloning and limited amplification of cDNAs in different size ranges, using a plasmid vector designed to facilitate transposon-mediated sequencing.
Ultimately, the purpose of the two most distinctive attributes of this approach—that is, (1) the serial and corresponding size fractionation of template (cytoplasmic mRNA) and product (double-stranded cDNAs), and (2) the separate cloning and (limited) amplification of cDNAs in different size ranges—is to maximize representation of transcripts, irrespective of length and abundance, in the final cDNA libraries. Because mRNA complexity is lower in a size-fractioned than in unfractionated RNA, there is greater likelihood for representation of rare transcripts in a library that contains cDNAs in the corresponding size-range than in a cDNA library derived from unfractionated mRNA. This difference is even further increased by separately cloning, electroporating, and propagating in bacteria (for limited amplification) cDNAs and clones, respectively, in different size-ranges. As a result, competition for cloning and amplification among cDNAs that differ significantly in length is eliminated, thus minimizing biases in representation of transcripts in the final library that might otherwise arise due to differences in transcript length.
It should be noted that the size-fractionation approach also presents certain disadvantages. The primary drawback is the fact that representation, in the final libraries, is limited to transcripts within the range encompassed by the mRNA size-fractions used as template for cDNA synthesis. To date, we have successfully derived full-length-enriched cDNA libraries from size-fractionated mRNA up to 7.0 kb in length. It has been our experience that libraries derived from size fractions containing transcripts in the 7.0- to 9.0-kb range are more likely to contain cDNAs derived from contaminating unprocessed nuclear transcripts, which compromises representation of bona fide full-length cDNAs.
We used this approach to construct 47 full-length-enriched cDNA libraries from size-fractionated cytoplasmic mRNA obtained from brain and eye tissue at multiple embryonic stages (upper heads at 9.5 to 10.5 dpc; brain and eyes at 12.5, 13.5, 14.5, 15.5, 16.5, 17.5, and 18.5 dpc), and from postnatal days 1, 5, and 15, of the C57BL/6 mouse strain. A complete list of the libraries constructed, with information on tissue and mRNA size fraction of origin, number of primary recombinants obtained, and total number of 5′ ESTs generated, is presented in Table 1.
A List of the 47 Full-length-Enriched NIH_BMAP cDNA Libraries With Associated Information Regarding Tissue and mRNA Size Fraction of Origin, Number of Primary Recombinants Obtained, and Total Number of 5′ESTs Generated
To assess the effectiveness of the procedures that we used for size fractionation of cytoplasmic mRNA and for size-selection of double-stranded cDNA, and thus demonstrate the quality of the full-length-enriched cDNA libraries generated with our approach, we first verified that cDNA size-ranges in these libraries do indeed correspond to those of the mRNA size fractions from which they originated. Plasmid DNA preparations from 16 libraries were linearized with a homing endonuclease (PI-SceI) and electrophoresed on an agarose gel along with a DNA size ladder. As shown in Figure 1, in all 16 libraries, cDNA sizes varied within the range of the mRNA size fraction used as template for their syntheses. It should be emphasized that shown in Figure 1 are linearized plasmid DNA preparations of entire libraries, which include the 1691-bp pYX-Asc I cloning vector.

Distribution of clone sizes in the size-fractionated cDNA libraries to assess the efficacy of the method used for size-fractionation of cytoplasmic mRNA and size selection of double-stranded cDNA. Linearized plasmid DNA from each of 16 size-fractionated libraries representing size fractions from 0.5 to 1 kb to 5 to 7 kb were electrophoresed and compared with a 1-kb standard ladder (New England BioLabs). Note that the clone sizes include the 1.7-kb pYX-Asc I vector.
Next, we investigated the correspondence between actual and expected lengths of 1274 cDNAs, by comparing the lengths of their complete sequences with the size ranges expected for the cDNAs in the library they originated. As shown in Table 2, this analysis revealed an overall correspondence that ranged from 50% to 100%. It is noteworthy, however, that this analysis also revealed that 70% to 90% of the sequences with lengths falling below the size range expected for clones in the library were shorter by no >15% of the minimum length in the respective range, thus documenting the effectiveness of this approach to generate libraries enriched for full-length cDNAs. A complete list of all sequences and corresponding libraries is available as Supplemental data.
Correspondence Between Actual and Expected Lengths of 1274 CDNAs
Here we report the sequence characterization of 1274 full-ORF-containing NIH_BMAP cDNAs that we identified in these libraries, all of which have been submitted to NCBI/MGC. It should be emphasized that all NIH_BMAP libraries were also contributed to the MGC program, while being characterized and sequenced in our laboratory. As a result, an additional 1019 full-ORF sequences were derived from these libraries by the MGC sequencing team.
Selection of Full-ORF-Containing cDNAs for Full-Insert Sequencing
The first step in the clone selection pipeline involved arraying of the cDNA libraries and production of 5′ ESTs. The number of 5′ ESTs derived from each library is listed in Table 1. In total, 175,990 5′ ESTs were generated and subjected to informatics analyses (sequence homology-based and ab-initio methods) for selection of full-ORF clone candidates. Alignment of the 175,990 5′ ESTs to the mouse genomic sequence using a dedicated BLAT server (Kent 2002; Karolchik et al. 2003) enabled the identification of 14,973 EST clusters. The 5′ most EST from each cluster was then identified and subjected to further analyses. These included (1) BLAST searches against RefSeq, Riken, SWISS-PROT, and MGC databases; (2) genomic context examination, based on data available in the UCSC database, to determine whether a 5′ EST overlaps with the start codon of a known gene or mRNA and whether it is the most 5′ of all ESTs mapping to that genomic location; and (3) ab initio tools for classification of 5′ ESTs according to the likelihood that they represent full-ORF-containing cDNAs. The latter are based on recognition of distinctive sequence features, such as the presence of a Kozak sequence motif and the occurrence and localization of start and stop codons, and decision tree optimization based on historic true/false-positive rates (http://genome.uiowa.edu/techreports.html). Selected clones were BLASTed against NCBI's “nr” database and GenBank records of each significant hit (e value < 10-8) were examined to seek homologous genes that had an annotated CDS for additional evidence that might further support or contradict the prediction.
A total of 7774 cDNA clones were selected according to these criteria as putative full-ORF-containing cDNAs. Of those, 3551 corresponded to transcription units already represented in the mouse MGC database and hence were not selected for full-insert sequencing, but still remain of interest due to their potential of representing alternatively spliced variants. Of the remaining 4223 clones, 3579 were rearrayed and 3′ ESTs were generated, and 644 await processing. Upon visual inspection and analysis by an annotator in the finishing group, 2084 clones were selected for in vitro transposition and full-insert sequencing, and the remaining 1495 clones were rejected.
Complete and accurate full-insert sequence has been obtained for 1863 clones, and 221 clones are currently in the finishing pipeline. Final analyses resulted in the classification of 1274 NIH_BMAP clones as full-ORF-containing cDNAs and in the rejection of 589 finished sequences for one of the following reasons: chimeric (5), frame shift (142), retained intron (113), library artifacts (22), unable to sequence (131), no significant ORF (39), 5′ truncation of ORF (54), 3′ truncation of ORF (30), a similar clone appeared in MGC while ours was still in the finishing phase (44), and 5′ end sequence of the re-arrayed clone did not match that of the original 5′ EST (9).
In conclusion, 52% (7774/14,973) of the cDNAs in the nonredundant set of clones that we identified in the NIH_BMAP cDNA libraries were ranked as full-ORF-containing candidates based on analyses performed on their 5′ ESTs. Of those, 72% to 81% (5635 to 6279/7774; 644 pending and 1495 rejected) remained considered as full-ORF-containing candidates upon further inspection and analysis of their 3′ ESTs. Finally, 68% (1274/1863) of the clones selected for full-insert sequencing, ultimately met all criteria required for classification as full-ORF-containing cDNAs.
Distribution of cDNA Size and ORF Length in NIH_BMAP Clones: Comparison to the Remainder Mouse MGC cDNAs
The distribution of cDNA sizes and ORF lengths in the 1274 NIH_BMAP cDNA sequences reported in this manuscript are presented in Figure 2, A and B, respectively. In addition, a complete list of the 1274 clones with their respective sequence and ORF lengths is available as Supplemental data to this manuscript. As shown in Figure 2A, although cDNA sizes range from 507 to 7146 bp, most sequences are in the 3.0- to 5.0-kb range (74%; 943/1274), with a peak of ∼4 kb (28%; 358/1274). On the other hand, as shown in Figure 2B, although ORF lengths range from 204 to 6849 bp, the majority fall within 1.0 to 3.0 kb (82%; 1048/1274), with a peak at ∼1 kb (38%; 481/1274).
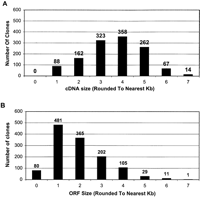
Size distribution of the 1274 full-ORF clones. For the 1274 full-ORF clones derived from NIH-BMAP libraries and sequenced locally: the distribution of insert sizes (A) and the distribution of ORF sizes (B).
A comparison of the size distribution of the NIH_BMAP cDNAs that are in the mouse MGC database with that of all other mouse MGC cDNAs revealed a striking difference, in that ∼90% of the cDNAs in the latter group fall in the 1.0- to 3.0-kb range, with a peak ∼2 kb (Fig. 3A). This is in sharp contrast with the size distribution of the NIH_BMAP cDNAs, with ∼75% of the sequences falling in the 3.0- to 5.0-kb range. This difference is particularly notable in the subset of MGC sequences derived from longer cDNAs (Fig. 3B), with NIH_BMAP clones composing 68% (493/727) of all mouse MGC clones ≥5 kb and 54% of those ≥4 kb (1060/1961). It is noteworthy that all NIH_BMAP cDNA sequences, comprising 20% of the MGC database, were included in this analysis irrespective of whether they were generated by our group at the University of Iowa or by laboratory members of the MGC sequencing team.
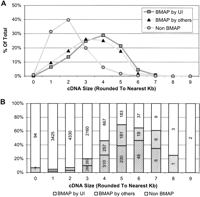
BMAP clones/sequences in the mouse MGC collection as of March 17, 2004. Three categories of clones are shown: 1077 BMAP clones sequenced by UI, 1019 BMAP clones sequenced by others, and non-BMAP clones (which represent all the clones in the MGC collection that are not BMAP clones). (A) Size distribution as a percentage of total clones within each category. (B) Percentage of clones contributed by each category, by clone size. The number of BMAP clones in MGC sequenced by UI from clone sizes 1 and 2 are 69 and 146, respectively. The number of BMAP clones in MGC sequenced by others for clone sizes 1 and 2 are 98 and 183, respectively.
NIH_BMAP cDNAs Predominantly Expressed in Brain and Eye Tissues
We have analyzed sequences derived from the 1274 full-ORF-containing NIH_BMAP cDNAs reported in this manuscript to identify those corresponding to transcripts most distinctively expressed in brain and eye tissues, respectively. We used NCBI's mouse UniGene database (build no. 135) to identify UniGene clusters containing sequences derived from the 1274 NIH_BMAP cDNAs. We then obtained information on the tissue of origin of every EST constituent of the 1274 clusters, using a locally curated translation file that specifies the tissue source(s) for each library indentification in the mouse UniGene, and determined the relative representations of ESTs derived from brain and eye cDNA libraries, respectively, in each cluster (shown as percentages in Table 3A,B). Next, we selected the NIH_BMAP transcripts in clusters with highest relative representations of brain and eye ESTs. Last, we determined, within each group, the subset of ESTs that originated from embryonic brain and embryonic eyes, respectively. This was calculated for each cluster, based on the ratio of the number of embryonic brain- (or embryonic eye-) ESTs per total number of brain (adult + embryonic) or eye (adult + embryonic) ESTs in the cluster (also shown as percentages in Table 3A,B).
Top 52 NIH_BMAP Transcripts Most Distinctively Expressed in the Brain
Top 50 NIH_BMAP Transcripts Most Distinctively Expressed in the Eye
It should be emphasized that only ESTs obtained from libraries derived exclusively from brain or eye tissues were counted as evidence for expression in the brain or in the eyes, respectively. Thus, this analysis provides a conservative estimate of transcript expression in these tissues.
A total of 3,089,497 ESTs in the mouse UniGene were used for these analyses, comprising 531,536 brain ESTs (17.2%), of which 216,120 are embryonic (7.0%), and 132,625 eye ESTs (4.3%), of which 27,721 are embryonic (0.9%).
Fifty-two NIH_BMAP transcripts expressed exclusively or predominantly in the brain, arbitrarily defined as those in UniGene clusters with >75% of ESTs derived from brain tissue, were identified in this analysis (Table 3A; Fig. 4). Included in this set are several transcripts known to be distinctively expressed in the brain, such as calneuron 1 (Wu et al. 2001); seizure-related gene 6 (Shimizu-Nishikawa et al. 1995); kinesin family member 1A (Okada et al. 1995); cerebellin 3 (Pang et al. 2000); T-box brain gene 1 (Bulfone et al. 1995); adenylate cyclase activating polypeptide 1 receptor 1 (Sheward et al. 1998); leucine-rich repeat LGI family, member 1 (Gu et al. 2002; Kalachikov et al. 2002); sulfotransferase family 4A, member 1 (Sakakibara et al. 2002); protocadherin 8 (Strehl et al. 1998); adenosine deaminase, RNA-specific, B2 (Mittaz et al. 1997); potassium inwardly-rectifying channel, subfamily J, member 9 (Lesage et al. 1994); and glutamate receptor, ionotropic AMPA1 GluR1 (Puckett et al. 1991). Interestingly, however, this analysis revealed several transcripts that are still uncharacterized and that seem to be mainly, but not highly, expressed in the brain (e.g., Mm.296323, Mm.337426, Mm.334249, Mm.334408). Yet a third class included uncharacterized transcripts apparently differentially and highly expressed in the brain (e.g., Mm.279818, Mm.44413, Mm.246605). In addition, we identified transcripts that seem to be expressed at relatively low levels, but specifically, in embryonic brain (e.g., Mm.334249, Mm.334408).
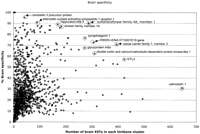
Graph of brain-specific versus EST composition. Gene identities were annotated for several of the most prevalent and most brain-specific clusters.
In contrast, only four of the 1274 NIH_BMAP transcripts were found in UniGene clusters containing ≥50% ESTs derived from eye tissue (Table 3B), of which three appear to be moderately or highly expressed in both embryonic and adult eyes (crystallin β A4, cyrstallin β A2, and dopachrome tautomerase). The fourth (Mm.246812), however, is a rare and yet uncharacterized embryonic eye transcript (BC067074) formerly, and only once, identified in a kidney cDNA library from a 14-month-old male mouse (BC042787). BLAT analysis indicates that the eye and kidney transcripts use different alternatively spliced 3′ terminal exons. This analysis also revealed a number of NIH_BMAP transcripts that are highly, yet not predominantly, expressed in the eye, some of which are known (e.g., Mm.1008, Mm.1860) and others are yet uncharacterized (e.g., Mm.55143). Several transcripts were identified in the 25% to 50% range of eye-specific expression, among which are a number of yet-uncharacterized transcripts expressed over a wide range in the eye.
In conclusion, based on the preliminary characterization of the 1274 NIH_BMAP full-ORF-containing cDNA sequences reported in this manuscript, we anticipate that these NIH_BMAP full-length-enriched cDNA libraries will prove invaluable not only for identification of additional full-ORF sequences derived from transcripts expressed in the developing mouse nervous system but also as a resource for identification of brain-specific transcripts, resulting from brain-specific splicing and/or polyadenylation, utilization of brain-specific promoters, as well as brain-specific antisense transcripts.
METHODS
Tissue Dissection
Time-pregnant C57BL/6 mice were purchased from either Charles River (Wilmington, MA) or Harlan (Indianapolis, IN). For embryonic days 9.5 to 10.5, the head was cut just anterior to the developing mandible and through the middle of the hindbrain. The developing eye was included in this cut. For embryonic days 12.5, 13.5, 14.5, 15.5, 16.5, 17.5, and 18.5 and postnatal days 1, 5, and 15, eyes and brains were dissected separately. The freshly dissected tissues were collected in DEPC-treated phosphate buffered saline and used immediately for the isolation of cytoplasmic RNA.
Isolation of Cytoplasmic mRNA
Cytoplasmic RNA was isolated essentially as described before (Favaloro et al. 1980). The tissue was homogenized in lysis buffer (140 mM NaCl, 1.5 mM MgCl2, 10 mMTris-HCl pH 8.6, 0.5 NP-40, and 10 mM vanadyl-ribonucleoside complexes) by using a tissue grinder (Kontes) with a loose pestle. Five milliliters of lysed tissue was then transferred to a 13 mL Sarstedt tube with 5 mL sucrose-containing lysis buffer (0.7 M sucrose) with 1% NP-40 and centrifuged in a Sorvall HB-6 rotor at 14,000g for 20 min at 4°C. The upper layer was carefully transferred to a Sarstedt tube containing one volume of 2× proteinase K buffer (20 mM Tris-HCl at ph 7.5, 10 mM EDTA, 1% SDS). Proteinase K was added to a final concentration of 200 μg/mL and incubated at 37°C for 30 min. The RNA was extracted with one volume of phenolchloroform, centrifuged in a Sorvall SS34 rotor at 12,000g for 20 min at 4°C and then precipitated with 2.5 volumes of ethanol and 0.1 volume of sodium acetate (pH 5.2). RNA samples were digested with 10 U RNase-free DNase (Roche) in 1 mM EDTA, 50 mM MgCl, and 250 mMTris HCl (pH 7.5) buffer for 30 min at 37°C, and poly(A)+ mRNA was purified using Oligotex mRNA kit (Qiagen) or Dynalbeads mRNA direct kit (Dynal).
Size Fractionation of poly(A)+ mRNA
Poly(A)+ mRNA (∼5 μg) was ethanol precipitated, resuspended in deionized formamide, and loaded on a 1% low melting temperature agarose gel, next to the 1-kb RNA ladder used as reference for size fractionation of the mRNA. The RNA ladder, but not the poly(A)+ mRNA, was stained with ethidium bromide and exposed to UV light to guide the otherwise blind size fractionation of the poly(A)+ mRNA. Gel slices containing poly(A)+ mRNA fractions of 0.5 to 1.0 kb, 1.0 to 2.0 kb, 2.0 to 3.0 kb, 3.0 to 4.0 kb, 4.0 to 5.0 kb, and 5.0 to 7.0 kb were melted at 65°C and digested with 0.3 U of Agarase (Promega) at 40°C.
Construction of cDNA Libraries From Size-Fractionated Cytoplasmic mRNA
cDNA libraries were constructed from each individual mRNA size fraction, essentially as we previously described (Bonaldo et al. 1996). Typically, each cDNA library was derived from 0.2 μg size-fractionated poly(A)+ mRNA. Briefly, first-strand cDNA synthesis was primed with a dT18 oligonucleotide containing a NotI site, for directional cloning, and a library-specific sequence-tag of 10 nucleotides positioned between the NotI site and the dT18 (Gavin et al. 2002), under conditions that we have optimized for generation of cDNAs with short dA/dT tails (Soares and Bonaldo 1998), in a reaction with 400 U reverse transcriptase (Superscript II, Life Technologies) and 0.5 mM each dATP, dTTP, dGTP, and methyl-dCTP, for 2 h at 37°C. Double-strand cDNA was synthesized by nick translation in a reaction with Escherichia coli DNA polymerase (New England Biolabs), E. coli DNA Ligase (New England Biolabs), and RNase H (USB), and size fractionated by agarose gel electrophoresis according to the size range of the mRNA size fraction used as template for first-strand cDNA synthesis. Size-selected cDNAs were ligated to EcoRI adaptors, digested with NotI, phosphorylated, and directionally cloned into the pYX-Asc I vector, doubly digested with EcoRI and NotI. The cDNA libraries were electroporated into phage (T1)-resistant-DH10B E. coli, and after 1 h at 37°C, 30% of the library was plated onto agar plates containing ampicillin, from which individual colonies were robotically picked and arrayed into 384-well plates. The remaining 70% was grown at 37°C overnight, and plasmid DNA was prepared by using a Qiagen kit.
We used the following library tags: (1) brain libraries: CAGCCACGAC (E18.5 dpc), GTGCGTGGAA (E15.5 dpc), TGAGAGAGCC (E12.5 dpc), AGCGAGACAG (pool of E13.5, 14.5, 16.5, and 17.5 dpc), CGAACTGAAT (E9.5 to 10.5 dpc), and CGAACTGAAT (pool of postnatal days 1, 5, and 15); (2) eye libraries: TTATTGAAGT (pool of E12.5, 13.5, and 14.5 dpc), CTGCGTCCTC (pool of E15.5,16.5,17.5, and18.5 dpc), and AATAATTACG (pool of postnatal days 1, 5, and 15).
pYX-Asc I is a 1691-bp plasmid that we derived from the pYX vector originally constructed and kindly provided by Dr. M.J. Brownstein (NIH). The modifications that we introduced in the pYX plasmid include the addition of an AscI site to the polylinker and the deletion of a region containing nonessential sequence. The latter modification was introduced after our observation of multiple transposon integrations within this region in the in vitro transposition reactions performed for transposon-facilitated sequencing. The resulting vector (pYX-Asc I) is thus ideal for transposon-facilitated sequencing because, with the exception of the short polylinker sequence, transposon integration into any sequence in the vector renders it a nonviable clone. Additional information on the pYX-Asc I vector, including its complete sequence, can be obtained at http://image.llnl.gov/image/html/vectors.shtml.
Transposon-Facilitated Sequencing and Finishing
The cDNA clones that were identified as full-ORF-containing candidates not yet represented in MGC were rearrayed and sequence-verified (both 5′ and 3′ end-sequences were obtained). Selected clones were colony-purified and grown individually for 15 h at 37°C, and their cultures were combined into pools. An aliquot of each culture was saved as a glycerol stock. Each pool consisted of seven to 16 clones with a combined size of 40 to 50 kb. Plasmid DNA from each pool was purified by using a Wizard Plus SV miniprep DNA purification system (Promega), and a sample of the purified plasmid was loaded to an agarose gel as a quality-control measure and to determine DNA quantity. Approximately 150 ng of purified DNA was used in a transposition reaction performed with a Template Generation System (Finnezymes) according to the manufacturer's instructions. One quarter of the transposition reaction product was electroporated into DH10B cells, incubated for 1 h at 37°C and plated on agar plates under appropriate antibiotic selection. A Genetix QBot was used to array 384 bacterial colonies obtained from each pool into 384-well microtiter plates. The 384-well plates were incubated overnight at 37°C, and double-stranded plasmid DNA templates were prepared from the resulting glycerol stocks by using a microwave-mediated cell lysis method (Marra et al. 1999).
Two parallel sequencing reactions were performed on each DNA template by using the ABI PRISM dRhodamine terminator cycle sequencing kit and primers that correspond to priming sites on the transposon element. Reaction products were electrophoresed on an ABI PRISM 3700 DNA Analyzer. Once the sequence reads were generated, the phred/phrap/consed package (Ewing et al. 1998; Gordon et al. 1998) was used to assign quality scores, to assemble the sequence reads, and to view and to edit the assemblies. For each pool, the 3′ and 5′ ESTs of the pooled clones were included in the assembly. Each pool's assembly typically included one contig for each clone. Each pool's assembly was split into assemblies that corresponded to the individual clones by using ace_splitter (http://genome.uiowa.edu/pubsoft/software.html). The assembly of each clone was viewed in Consed to determine the overall error rate and to identify low quality regions and regions covered in only one direction.
All full-insert cDNA sequences were finished according to the following quality criteria: no gaps; no ambiguous bases (Ns); cumulative average phrap score of at least 40 (error rate not to exceed one error in 10,000 bases), and a phrap score of at least 30 for each individual base in the assembly, <5% single stranded coverage, and, for those rare single-stranded covered regions, at least three reads of coverage, or two reads of coverage and a phrap score of at least 40 for each base. Clones that did not meet these standards were finished via directed sequencing by using custom oligonucleotides. The oligos were designed in consed and ordered from Integrated DNA Technologies, Inc. The sequence reads generated with the oligos were added to the assemblies of the reads derived from the respective clones until the quality standards were met. The sequence of all finished clones was translated and aligned to known sequences by using BLAT and BLAST to identify possible problems (frame shifts, retained introns, deletions, substitutions). Clones that appeared to have a full intact ORF were submitted to MGC as full-ORF clones. All other clones were submitted to GenBank as full-insert sequences. It is noteworthy that this sequencing pipeline also tracks the status of each clone, all oligos ordered and final sequences of each clone in an easy to use graphical format.
Acknowledgments
We would like to thank Dr. Michael Brownstein (National Institute of Mental Health, NIH) for kindly providing the pYX plasmid. We also thank Dr. Steven O. Moldin (National Institute of Mental Health, NIH), Dr. Hemin Chin (National Eye Institute, NIH), and Dr. Robert Strausberg (while director of the Cancer Genomics Office at the National Cancer Institute, NIH, currently vice president for research at The Institute for Genomic Research [TIGR]) for facilitating the coordination between this component of the NIH-Mouse BMAP and the NIH-MGC Program. Dr. Bair's work was supported by an NRSA post-doctoral fellowship no. 1F32HG002881-01A1. V.C.S. is an investigator of the Howard Hughes Medical Institute. This work was supported by contract no. N01MH12006 to the University of Iowa (M. Bento Soares, principal investigator), entitled “Gene Discovery in the Developing Nervous System.”
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2601304.
-
↵11 Corresponding author. E-MAIL bento-soares{at}uiowa.edu; FAX (319) 335-9565.
-
- Accepted April 27, 2004.
- Received March 29, 2004.
- Cold Spring Harbor Laboratory Press








