
From ORFeomes to Protein Interaction Maps in Viruses
Abstract
Although cloned viral ORFeomes are particularly well suited for genome-wide interaction mapping due to the limited size of viral genomes, only a few such studies have been published. Here, we summarize virus interaction mapping projects involving vaccinia virus, hepatitis C virus (HCV), potato virus A (PVA), pea seed-borne mosaic virus (PSbMV), and bacteriophage T7, as well as some projects in progress. The studies reported suggest that virus-specific coding and replication strategies must be taken into account to yield accurate numbers of protein interactions. In particular, the number of false negatives can be significant for RNA viruses expressing precursor polyproteins (because interactions between full-length mature proteins are often not detected due to incorrect processing) and for viruses replicating in the cytoplasm whose transcripts have not been selected for splicing signals. In conclusion, the studies on viral protein interaction maps suggest that cloned pathogen ORFeomes will contribute to a holistic picture of the pathogenesis of infectious diseases and are ideal starting points for new approaches in systems biology. Both viral ORFeome and interaction mapping projects are being documented on our Web site (http://itgmv1.fzk.de/www/itg/uetz/virus/).
Completely sequenced genomes revealed that the parts lists of organisms are finite. This fact turns seemingly infinitely complex organisms into feasible research projects. Although this is true for both eukaryotes and prokaryotes, it is even more so for viruses. Viruses are important pathogens for humans, plants, and livestock, but there are few comprehensive studies tackling all genes or proteins of a single virus genome. However, this is changing rapidly, especially with the availability of cloned ORFeomes, that is, complete sets of plasmids or PCR products containing all open reading frames (ORFs) of a genome (Rual et al. 2004).
ORFeomes allow the systematic cloning and subsequent analysis of protein function by expressing the ORFs in suitable bacterial or eukaryotic systems. The proteins can be purified and analyzed biochemically or expressed in a cell, and their functions can be studied in a natural environment. Eventually all proteins must be studied in a variety of ways in order to gain a complete understanding of their physiological roles. Important parameters include their expression levels and their stability, their localization, interactions, and biochemical activities (attempts to integrate such data are discussed by Bader et al. 2003). Once we know these static parameters we can address their dynamics, that is, how they change during the life cycle of the virus or during different processes within the host, such as replication. To our knowledge, systematic studies on whole viral genomes have been focusing on protein interactions, with only a few other functional studies. In this review we primarily summarize viral protein interaction mapping projects and the lessons we have learned from them.
Sequenced Virus Genomes and ORFeomes
Although well over 1000 viral genomes have been completely sequenced (NCBI viral genomes Web site, http://www.ncbi.nlm.nih.gov:80/genomes/VIRUSES/viruses.html) there have not been many reports about cloning complete ORFeomes. Some cloned ORFeomes have been published in conjunction with some functional analyses, but it appears that most researchers still focus on individual proteins rather than complete proteomes. The exception may be the analysis of protein interaction maps, because they are usually done using the yeast two-hybrid system which requires either a random library made from genomic fragments or a defined clone set of all ORFs (Fig. 1). Complete ORF clone sets are perfectly suited for systematic analysis of protein interactomes, because every possible protein combination can be tested.
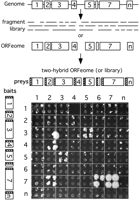
From viral ORFeome to protein interaction map. ORFs can be predicted from sequenced genomes with high confidence and subsequently cloned. Alternatively, libraries of random fragments can be generated. ORFs or fragments can then be cloned into two-hybrid vectors and tested for pairwise interactions in an array format (Uetz 2002). In this experiment, ORFs from KSHV were tested for interactions. Note that each test was done in quadruplicate to ensure reproducibility. Interactions between bait 3 and prey 5 (3-5) is also reproducible in the inverted configuration (bait 5 and prey 3), whereas interactions 4-2 and 7-2, and 7-6 are not. ORF 7 is homodimerizing (7-7). Colony 3-2 is a clear false positive as it is not reproducible in quadruplicate; 4-2 and 7-6 are considered true positives, although only three of four colonies are growing, which may be due to imprecise spotting. Also shown is the noticeable background growth, which can be easily distinguished from the two-hybrid signal in this array format.
As new viral ORFeomes are expected to become increasingly available, we have set up a Web site that reports such ORFeomes. We solicit authors and companies who offer such clone collections to submit relevant information for inclusion on this site (http://itgmv1.fzk.de/www/itg/uetz/virus/).
Generating Protein Interaction Maps of Viruses
Specialized databases for protein-protein interactions contain surprisingly little data on viruses. At the time of this writing (March 2004), the Database of Interacting Proteins (http://dip.doe-mbi.ucla.edu/) contained only about 100 interactions involving viral proteins (Salwinski et al. 2004). The number was even smaller in the Biomolecular Interaction Network Database (BIND; http://www.bind.ca). Obviously, only a small number of viruses has been studied by systematic two-hybrid analysis (Uetz and Hughes 2000). Usually these maps have been generated by systematic two-hybrid testing of ORF pairs for interactions, typically in an array format (Uetz 2002). Although two-hybrid results from array screens are usually fairly clear, some interactions suffer from background due to nonspecific interactions of the bait (Fig. 1). Thus, it is advisable to verify a two-hybrid interaction by some independent method. Many authors have used GST pull-down experiments or coimmunoprecipitation for this purpose.
Surprisingly, only two larger viruses have been systematically analyzed for protein interactions, namely Vaccinia virus (McCraith et al. 2000) and Kaposi sarcoma associated herpesvirus (KSHV; P. Uetz, Y.A. Dong, C. Zeretzke, M. Roupelieva, D. Rose, C. Atzler, and J. Haas, in prep.). As viral ORFeomes can be easily cloned, and the number of two-hybrid assays is rather limited (102 assays for 10 proteins, 104 assays for 100 proteins), we expect many more such studies in the near future.
False Negatives: How Comprehensive Are Two-Hybrid Maps?
Numerous two-hybrid studies showed that the two-hybrid system never detects all physiological interactions. However, this limitation is also true for any other method. Aloy and Russell (2002) showed that the two-hybrid system tends to detect transient interactions, whereas interactions within protein complexes are more efficiently detected using purified complexes in combination with mass spectrometric analysis. No matter which proteins are analyzed, the two-hybrid system has a significant number of false negatives; that is, true biological interactions are not detected by the system. Edwards et al. (2002) estimated the number of false negatives in two-hybrid screens to be between 43% and 71%, based on a comparison of two-hybrid data and the crystal structures of the proteasome and the Arp2/3 complexes, whose protein pairs were systematically tested by two-hybrid analysis. The false-negative rates in genome-wide screens are usually even higher, because each pair cannot be tested repeatedly.
False Positives: How Reliable Are Two-Hybrid Maps?
Several studies suggest that genetic interaction networks assessed by two-hybrid screens can be fairly reliable. In the study by Li and colleagues (2004) on the interactome network of Caenorhabditis elegans, between 35% and 82% of the interactions found by large-scale two-hybrid screens were confirmed by coaffinity purification assays. In KSHV, ∼50% of the protein interactions could be confirmed by coimmunoprecipitation, suggesting the accuracy of the two-hybrid results (P. Uetz, Y.A. Dong, C. Zeretzke, M. Roupelieva, D. Rose, C. Atzler, and J. Haas, in prep.). The failure of coimmunoprecipitation does not necessarily indicate that the two-hybrid results are false positive, as structural constraints might play a different role in coimmunoprecipitation. The number of false positives can also be reduced by statistical means. Giot and colleagues (2003) used an automated confidence scoring algorithm to reduce the number of false positive interactions in a protein interaction map of Drosophila.
Lessons Learned From Interaction Maps
Protein interaction data should be interesting to most virologists, but especially to structural biologists. Interaction maps provide a mental map for the intricate relationships among proteins and suggest hypotheses about their assembly, regulation, modifications, etc. However, the biological consequences are beyond the scope of this article and have been summarized elsewhere (Titz et al. 2004). Here we will highlight some technical and biological lessons learned specifically from whole-genome analyses of viral proteomes.
RNA Viruses: HCV, WSMV, PVA
Many RNA viruses express a precursor polyprotein which is cleaved into several mature viral proteins either autocatalytically or by cellular proteases. The translation of the precursor polyprotein and its cleavage is mandatory for correct folding and processing of the mature proteins, and individually expressed mature proteins may not be correctly processed (Flajolet et al. 2000).
Not surprisingly, such mature proteins have not been tested very successfully for pairwise interactions. In both hepatitis C virus (HCV) and wheat streak mosaic virus (WSMV), no interactions between mature proteins have been detected by two-hybrid assays (Choi et al. 2000; Flajolet et al. 2000). In contrast, when random fragments were tested, a number of interactions could be detected. Besides incorrect processing, an alternative explanation for this observation might be that only certain protein fragments can productively act in conjunction with a fused activation or DNA-binding domain, with the consequence that the interaction can only be detected if these fragments are used. In WSMV, which belongs to the filamentous potyvirus (+) strand RNA virus family, the polyprotein precursor is cleaved into nine mature proteins. When Choi et al. (2000) tested the mature proteins by two-hybrid assays, they could not detect any heterologous interactions. In contrast, highly promiscuous interactions are found when random fragments were used: the proteins P1, HC-Pro, P3, and CI interacted with each other in all possible combinations. Interactions of the P3 protein were only found in in vitro assays, not in two-hybrid tests. Similar observations have been made in HCV, which belongs to the flavivirus (+) strand RNA virus family: Full-length proteins did not result in any interactions when tested by two-hybrid assays (Flajolet et al. 2000). However, screening of random fragments resulted in five interactions, such as the known interaction between the capsid homodimer and the protease dimer and between the nonstructural proteins NS3 and NS4a, as well as several novel interactions such as between NS2 and NS4a.
However, polyprotein expression and processing is not necessary for all interactions to be detected among processed RNA virus proteins. In both potato virus A (PVA) and pea seed-borne mosaic virus (PsbMV), which belong to the potyvirus (+) strand RNA virus family similar to WSMV (Guo et al. 2001), as well as in a subset of poliovirus proteins (Cuconati et al. 1998), interactions have been detected among cloned mature proteins.
These studies indicate that due to the specific features of RNA virus translation, both fragment and full-length approaches should be combined in such viruses for a more complete catalog of protein-protein interactions.
Bacteriophage T7
The first genome-wide two-hybrid study was carried out on bacteriophage T7. Bartel et al. (1996) screened a library of random T7 protein fragments and tested cloned ORFs against each other. Among the 55 phage proteins, they found 25 interactions, including four interactions that had been described previously (Fig. 2). Unfortunately, as not all ORFs were cloned and tested as full-length ORFs, it is difficult to compare the two data sets.
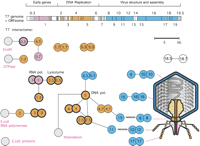
ORFeome and protein interaction map in bacteriophage T7. Cloned ORFs and random fragments were used to generate a two-dimensional interaction map. ORFeomes can also be used by structural genomics projects to derive 3D structures. A combination of interaction maps and crystal structures often allows the reconstruction of protein complexes as in viral particles or their subunits. The top panel shows the T7 genome with each ORF represented as a box. Note that only the main ORFs are indicated by integral numbers as originally identified by genetic screens. Genes in between have decimal numbers. Proteins that have been found to interact with others are indicated by colored boxes; box colors indicate a simplified assignment to functional classes as shown by the heading. Interactions between proteins are shown as lines, with self-interactions shown as dimers. Hatched lines indicate expected interactions which have not been found by two-hybrid analysis. Gray proteins correspond to host proteins (E. coli). Blue proteins are involved in virus assembly and structure, and their rough location in the virus particle is shown on the right, if known. Proteins with thick borders have been crystalized and their structure is known. Genetic map modified after Dunn and Studier (1983). Interactions primarily based on data from Bartel et al. (1996).
Vaccinia and KSHV: Comparing Methods and Results
Vaccinia virus and Kaposi sarcoma-associated herpesvirus (KSHV) are thus far the largest viruses analyzed in a systematic way for protein interactions (McCraith et al. 2000). Both viruses are large double-stranded DNA viruses, but belong to different virus families with distinct biological features. Vaccina virus is a member of the poxvirus family, and KSHV belongs to the herpesvirus family. Intriguingly, despite their similar approaches, the analysis of the two viruses resulted in quite different numbers of protein-protein interactions.
In the study on vaccinia virus (McCraith et al. 2000), 266 ORFs were tested in all pairwise combinations by two-hybrid assays. The screen of about 70,000 protein pairs revealed only 37 interactions of which 13 (35%) were self-interactions (i.e., homodimers or homomultimers). Of the 24 remaining interactions, five (i.e., 13.5% of 37) were observed in both orientations, that is, with the two proteins as both bait and prey construct (cf. Fig. 1). This suggests that two-hybrid arrays should always include all possible combinations with every protein as both bait and prey (for a proteome of N proteins this requires N2 combinations and not just N2/2). Double-checking each pair not only verifies each combination but also avoids problems if one of the two partners is a transcriptional activator and therefore cannot be used as a bait. One of the reasons for the small number of interactions detected by McCraith et al. (2000) is probably caused by the fact that vaccinia virus replication and transcription take place in the cytoplasm. Thus, viral splicing signals have not been eliminated in the course of evolution. If these sequences are expressed in the nucleus of eukaryotic cells from transfected plasmids, a considerable number of transcripts may be artificially spliced and the proteins encoded either disrupted or structurally altered. In fact, we found 282 putative introns in the Vaccinia genome, that is, more than one putative intron per ORF (predictions based on http://www.cse.ucsc.edu/~leslie/runIntronSearch.html). Another drawback of the vaccinia study might be that transmembrane proteins were only expressed as full-length proteins. Because the two-hybrid system is based on an interaction of bait and prey fusion proteins in the nucleus, this may have contributed to the large number of interactions not detected by McCraith et al. (2000).
KSHV belongs to the γ herpesvirus subfamily similar to Epstein-Barr virus (EBV) and encodes ∼88 proteins (the exact number is currently unknown, because some as yet uncharacterized viral proteins might be generated by splicing of short ORFs not known to be expressed). Herpesviral replication, transcription, and capsid assembly occur in the nucleus, and herpesviral transcripts can be spliced similarly to host mRNAs. An array-based screen of all protein pairs revealed 125 protein interactions (P. Uetz, Y.A. Dong, C. Zeretzke, M. Roupelieva, D. Rose, C. Atzler, and J. Haas, in prep.). Several protein interactions have been shown before, for example the interaction between the two capsid proteins ORF 25 and ORF 65, and the protein interaction between the immediate early transcriptional activators ORF 57 (RTA) and K8. In addition, a reasonable number of protein interactions have been predicted, for example the interaction between different DNA packaging and tegument proteins. A surprisingly large number of protein interactions, however, has not been shown before and occur between viral proteins which were previously thought to belong to different functional groups.
In summary, our analysis of KSHV shows that, probably due to the spacial and coding constraints in viruses, (1) a complex protein network exists, in which proteins of distinct functions are involved, (2) a considerable number of herpesviral proteins probably have multiple functions, and (3) several previously unidentified proteins possess a large number of interaction partners (in networks called “hubs”) and probably exert a prominent, coordinative function in viral pathogenesis (Fig. 3). Our study of KSHV also indicates that protein interactions in herpesviruses constitute a scale-free network and that interacting proteins are more conserved, similar to protein interaction networks of other organisms. Finally, interacting proteins tend to be coexpressed. In comparison to vaccinia virus, we found a considerably larger number of protein interaction in KSHV (Table 1). As indicated above, this could be caused by the differences in viral replication and transcription strategies. In addition, technical differences such as the expression levels of the two-hybrid vectors used may account for the discrepant findings in these studies.
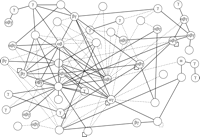
Protein interaction map of Kaposi's sarcoma associated virus (KSHV), highlighting conserved interactions and data quality. The protein interaction map of KSHV involves roughly half of all KSHV proteins. We assume that the other proteins interact with host proteins or their interactions have not yet been identified. Protein conservation is indicated by Greek letters, namely that the protein is conserved in α herpesviruses (HSV1), β herpesviruses (CMV), and/or γ herpesviruses (EBV). Solid lines indicate interactions that have been confirmed by coimmunoprecipitation; hatched lines were found by two-hybrid only. Note that only three of these interactions have been published thus far; the remaining ones are derived from a recent genome-wide two-hybrid screen (from P. Uetz, Y.A. Dong, C. Zeretzke, M. Roupelieva, D. Rose, C. Atzler, and J. Haas, in prep.). Interestingly, the proteins in this map are much better connected than in the T7 map (Fig. 2), and interactions of conserved proteins appear to be more reproducible than those of KSHV-specific proteins.
ORFeome and Interaction Studies of Viral Proteomes
Outlook
With the help of pathogen ORFeomes, a plethora of new data sets can be generated which will allow new views on the pathogenesis of infectious diseases, but pathogen-specific features must be taken into account to perform these genome-wide screens as usefully and accurately as possible. Aside from protein interaction mapping, virus ORFeomes have barely been used for other proteomic investigations. For example, ORFeomes could be used for a systematic analysis of virus-host interactions and protein localizations (as GFP fusions), structural genomics, or systematic functional analysis (e.g., using biochemical activities or mutagenesis; Martzen et al. 1999; Yu et al. 2003).
Furthermore, the increasing number of genome sequences from pro- and eukaryotic species will allow us to study host-virus interactions in much more detail. We expect two-hybrid arrays and whole-proteome chips of humans and other host species (e.g., Zhu et al. 2001) that can be screened with all proteins of their corresponding viruses. This would provide, for the first time, comprehensive insight into the interactions of host and pathogen proteins.
The number of eukaryotic viruses and phages is probably considerably higher than previously thought. Rohwer (2003) estimated that only 0.0002% of the global phage metagenome has been sampled. Similarly, in a number of new phages that have been sequenced recently, 50% of the ORFs in these genomes are unrelated to any sequence in GenBank (Pedulla et al. 2003). Given that only a tiny subset of all sequenced viral proteomes has been analyzed for protein interactions, we get a sense of how much work remains to be done just to get a glimpse of the variety of protein interactions in phage and other viruses. We believe that ORFeome-based techniques will provide us with a better understanding of viral infections which will, eventually, allow us to improve our strategies to cure them.
Footnotes
-
[Supplemental material is available online at www.genome.org. The interaction data from this study have been submitted to BIND under accession nos. 130817-130825, 130863-130879, 133397, and 133607.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2583304.
-
↵3 Corresponding authors. E-MAIL peter.uetz{at}itg.fzk.de; FAX +49 7247 82 3354. E-MAIL haas{at}lmb.uni-muenchen.de; FAX +49 89 5160 5292.
-
- Accepted May 21, 2004.
- Received March 16, 2004.
- Cold Spring Harbor Laboratory Press








