
Transcriptional Response of Lymphoblastoid Cells to Ionizing Radiation
Abstract
The effects of ionizing radiation (IR) on the temporal transcriptional response of lymphoblastoid cells were investigated in this study. We used oligonucleotide microarrays to assess mRNA levels of genes in lymphoblastoid cells at various time points within 24 h following γ-irradiation. We identified 319 and 816 IR-responsive genes following 3 Gy and 10 Gy of IR exposure, respectively, with 126 genes in common between the two doses. A high percentage of IR-responsive genes are involved in the control of cell cycle, cell death, DNA repair, DNA metabolism, and RNA processing. We determined the temporal expression profiles of the IR-responsive genes and assessed effects of IR dose on this temporal pattern of expression. By combining dose-response data with temporal profiles of expression, we have identified sets of coordinately responding genes. Through a genomic approach, we characterized a set of genes that are implicated in cellular adaptation to IR stress. These findings will allow a better understanding of complex processes such as radiation-induced carcinogenesis and the development of biomarkers for radiation exposure.
Ionizing radiation (IR) is used extensively in medical diagnostic and treatment protocols. It is also present at low levels throughout the environment. IR poses a major threat to cells by compromising genomic integrity and cellular viability. The increased risk for developing malignancies associated with IR exposure has been well documented (for review, see Bast and Gansler 2000). Even though precautions are taken to minimize exposure of healthy tissues to radiation, medical IR can still confer an increased risk of carcinogenesis to patients.
The manner by which IR damages cells is dependent on the type of radiation. Electromagnetic radiation ionizes cellular components indirectly through the generation of highly reactive free radicals, whereas protons and other heavy particles are direct ionizing agents. The major consequence of IR exposure is the generation of single or double-stranded breaks in DNA, which result in a cascade of events involving a complex network of signal transduction and transcriptional regulation. Damage to DNA elicits a cellular stress response that includes DNA damage recognition and cell cycle arrest, followed by DNA repair or apoptosis. If any of these processes fail, mutations can accumulate in the genome, resulting in malignant transformation of somatic cells or heritable mutations in germ cells.
Previous studies have utilized microarrays to describe gene expression changes associated with IR stress (Amundson et al. 1999, 2000, 2003; Khodarev et al. 2001; Tusher et al. 2001). However, little is known about the temporal pattern of gene expression following IR exposure in normal tissue and how IR dose affects these transcriptional changes. In this study, we used microarrays to conduct a genome-wide survey of the temporal transcriptional response of lymphoblastoid cells to IR exposure. We have identified genes that are induced or repressed following irradiation and have characterized their temporal expression profiles. Furthermore, we have examined the effects of IR dose on these IR-responsive genes. Identification and characterization of IR-responsive genes allow us to begin to understand the molecular mechanisms that underlie the pleiotropic effects of IR.
RESULTS
Transcriptional Profile Analysis of Lymphoblastoid Cells Exposed to 3 GY and 10 GY of Ionizing Radiation
We irradiated lymphoblastoid cells from 10 unrelated individuals at 2 different doses, 3 Gy and 10 Gy. Cells were harvested prior to irradiation (0 h) and at 1, 2, 6, 12, and 24 h after IR exposure. For each time point and dose, RNA was extracted from each cell line and the samples from all 10 individuals were pooled. A separate reference sample consisting of unirradiated lymphoblastoid cells from a different set of 10 individuals was also prepared. Pooled RNA samples were labeled and hybridized onto Affymetrix Human GeneChips (U95A).
Results from the 3 Gy and 10 Gy experiments were analyzed separately. For each gene, changes in expression levels were calculated by comparing the signal intensity of the irradiated sample with that of the unirradiated reference sample. Genes were considered IR responsive if their expression levels postirradiation were significantly different from the reference baseline as determined by the criteria modeled by d-Chip (Li and Wong 2001).
To assess the reproducibility of the microarray-based expression analysis, we compared the results from three independent hybridizations of the same RNA sample onto U95A GeneChip arrays. Labeling reactions and hybridizations were performed separately. The results from the three hybridizations were highly reproducible. By the same criteria used for analyzing the experimental data, only 11 genes of the total 12,559 genes displayed expression level differences in at least 1 of the 3 possible pair-wise comparisons. Of these genes, only 1 gene displayed greater than twofold expression difference between replicates.
IR-Responsive Genes
A total of 319 genes at the 3 Gy dose and 816 genes at the 10 Gy dose displayed changes in expression level compared with baseline (see Supplementary Material, available online at www.genome.org, for complete list). This represents 2.5% and 6.6% of all of the genes on the array, respectively. At the 3 Gy dose, 227 genes were repressed, 87 genes were induced, and 5 genes showed changes in both directions at various time points during the time course. Expression changes ranged from 4.5-fold repression to 5.3-fold induction. In contrast, at the 10 Gy dose, 156 genes were repressed and 660 genes were induced, ranging from 9.3-fold repression to 5.7-fold induction.
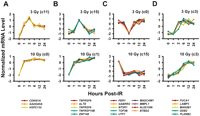
We validated a group of genes by quantitative RT–PCR using a different set of cell lines than those used in the microarray study (Fig. 1). In all cases, the quantitative RT–PCR results confirmed the U95A GeneChip data.
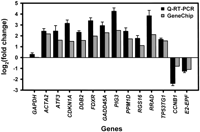
Comparison of U95A GeneChip data (gray bars) to quantitative RT–PCR (black bars). The quantitative RT–PCR values are means and standard errors for changes in expression levels (n = 8) at 12 h postirradiation following 10 Gy of IR exposure. GAPDH was used as a negative control. All genes were normalized to β-actin levels. U95A GeneChip values are derived from 10 individual cell lines pooled together at the same experimental conditions.
Functional Characteristics of IR-Responsive Genes
We classified the IR-responsive genes by assigning them to functional groups on the basis of Gene Ontology Consortium (GO) biological process categories (Ashburner et al. 2000; Fig. 2). For comparison, the non-IR-responsive genes on the U95A GeneChip array were also grouped by the same categories. We compared IR-responsive genes at each dose with the non-IR responsive genes on the array by Pearson's χ2 test of independence. Functional information is available for 52% of the genes on the array, and only these genes were included in the analysis. When compared with the non-IR-responsive genes on the array, a significantly larger proportion of IR-responsive genes following 3 Gy of IR exposure are functionally related to cell cycle, DNA metabolism, DNA damage/repair, and RNA processing pathways (P < 0.05 for each comparison after correcting for multiple testing). Similarly, following 10 Gy of IR treatment, a significantly larger proportion of IR-responsive genes are functionally related to cell death and DNA damage/repair pathways.
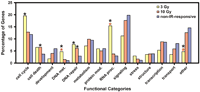
Functional grouping of IR-responsive genes. A black asterisk indicates categories significantly different from the non-IR-responsive genes on the U95A GeneChip (P <0.05 for each comparison after correcting for multiple testing). A red asterisk indicates categories significantly different between IR-responsive genes for 3 Gy and 10 Gy doses. Only genes with GO designations were used in the analysis. The standard error of the percentage of genes ranges from 1.0% to 3.0% for 3 Gy and 0.6% to 1.5% for 10 Gy.
IR-Responsive Genes in Common Between 3 GY and 10 GY IR Exposure
There are 126 IR-responsive genes in common between the 3 Gy and 10 Gy experiments (Table 1). These genes include a number of p53-dependent genes, general stress response genes, and cell cycle-related genes. Previous reports have shown that p53 plays a key role in response to cellular stress (for review, see Sharpless and DePinho 2002). In our 3 Gy and 10 Gy experiments, we identified several IR-responsive genes that are known to be p53 dependent. These include CDKN1A (p21), GADD45A, and DDB2, which play important roles in cell cycle arrest and DNA repair. Two p53-dependent death receptor genes, TNFRSF6 (Fas/APO-1) and TNFRSF10B (KILLER/DR5), were also induced shortly following IR exposure. Similarly, oxidative stress genes involved in p53-mediated apoptosis, such as PIG3 and FDXR, were up-regulated. Recent reports indicate PIG3 acts to stabilize p53, whereas FDXR generates oxidative stress in mitochondria (Asher et al. 2001; Hwang et al. 2001). Additionally, genes involved in general stress adaptation were also induced at both doses of IR exposure. Some examples include HSPCB, HSPE1, ATF3, and PPM1D. HSPCB and HSPE1 encode heat shock proteins, whereas ATF3 and PPM1D are downstream targets of MAP kinase signaling pathways. Many cyclins (CCNB1, CCNG1, CCNA2) and cell cycle-related genes (CDC20, CHC1, MCM6) displayed gene expression changes as well.
IR-Responsive Genes in Common for Both 3 Gy and 10 Gy of IR Exposure
IR-Responsive Genes Specific for 3 GY Or 10 GY IR Exposure
A large number of genes appear to respond specifically to only one of the IR doses. The higher radiation dose elicited transcriptional changes in a larger number of genes. The greater degree of insult caused by a higher radiation dose triggers a more complex response.
Specifically, 10 Gy of IR induced a number of DNA repair genes, including BLM, ERCC4, NBS1, RAD51C, and XPC, which are not affected at the 3 Gy dose. In addition, the p53-regulated genes, MDM2 and PCNA, displayed increased expression levels. A higher radiation dose also activated many cell death-related genes, including a large group of anti-apoptotic genes (BAG2, BCL2, BCL2A1, BCL2L2, and BNIP3). Although interferon transcripts did not show detectable changes in levels, several transcripts for interferon-inducible proteins along with those for interferon α and γ receptors showed elevated expression levels by 24 h postirradiation. MAP kinase-associated genes, such as MAP2K4, MAP2K6, MAP3K5, and MAP3K7IP2, were also induced at later time points during the 24-h time course. Finally, the higher radiation dose caused increased transcript levels of a group of oxidative stress genes, including ATX1, OSR1, NFE2L2, GPX1, and GPX4.
In contrast, a large number of RNA processing/modification genes show gene expression changes specifically at the 3 Gy dose. These include hnRNPs (HNRPC, HNRPD, HNRPH3, HNRPM, HNRPR, and HNRPU) and splicing factors (SFRS1, SFRS3, SFRS6, and SFRS10), which display a gradual decline in transcript levels through the 24-h period following IR exposure.
Irradiation Dose-Dependent Gene Expression Patterns
To understand the effects of IR dose on temporal expression patterns of IR-responsive genes, we focused on the 126 genes that responded to both doses of IR and compared their expression profiles. For each of these IR-responsive genes, the correlation coefficient between the 3 Gy and 10 Gy expression profiles was calculated.
Figure 3A shows that the temporal pattern of expression for certain genes remains quite similar between the two IR doses. The 10 Gy IR dose caused a larger magnitude of expression change for some of the genes, whereas other genes showed identical transcriptional profiles following either IR dose. Most of these genes with similar profiles at both doses are late IR-responsive genes, which exhibit gradual induction or repression kinetics.
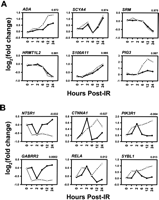
Genes with the most similar (A) and the least similar (B) expression profiles between the 3 Gy (solid line) and 10 Gy (broken line) doses. For each gene, the correlation coefficients between the expression profiles are indicated at the top, right.
Conversely, there are genes that display extremely different temporal expression patterns at varied IR doses (Fig. 3B). The majority of these genes are early responders to IR damage. At the 3 Gy dose, these IR-responsive genes exhibit transient expression changes that peak at 2 h postirradiation. In contrast, at the 10 Gy dose, these genes exhibit more rapid gene expression changes (occurring at 1 h postirradiation) that last longer in duration. Experiments using quantitative RT–PCR showed that return to basal expression levels of some genes is reached after ∼72 h postirradiation (data not shown).
Coordinated Expression Profiles of IR-Responsive Genes
Although some IR-responsive genes display different temporal expression patterns depending on the dose of IR exposure, there are groups of genes that show very similar temporal expression patterns relative to each other at both the 3 Gy and 10 Gy IR doses. These genes are of interest, as this similarity may imply that related pathways regulate these groups of IR-responsive genes.
To identify sets of genes that are highly correlated in their expression patterns at both doses, we grouped the 126 IR-responsive genes that are in common between the doses by similarity in their temporal expression patterns using GeneCluster 2.0 (Golub et al. 1999; Tamayo et al. 1999). This program creates gene clusters by using self-organizing map (SOM) algorithms. The IR-responsive genes were grouped into 16 clusters at each dose, generating sets of genes with distinct patterns (Fig. 4). Then, we searched specifically for groups of genes that clustered together at both 3 Gy and 10 Gy IR doses.
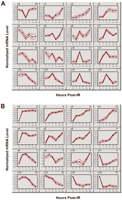
Gene expression patterns for the 126 IR-responsive genes in common between 3 Gy (A) and 10 Gy (B) doses. Clusters were generated using self-organizing maps (GeneCluster 2.0). Clusters for each dose were calculated separately. For each cluster, the cluster number is indicated at the top, left, and the number of genes assigned to each cluster is indicated at the top, right. Graphs for each cluster show cluster means (black line) flanked by standard deviations (red lines). Each gene was normalized across time points to have mean = 0 and SD = 1.
Several groups of known coregulated genes were found (Fig. 5). For example, the known p53-regulated genes, CDNK1A and GADD45A, clustered together in both the 3 Gy and 10 Gy experiments (Fig. 5A). Both of these genes are important downstream effectors of the p53 pathway and are involved in similar pathways. The gene for a hypothetical protein HSPC132 also clusters with CDNK1A and GADD45A, which suggests that HSPC132 may be regulated by p53, and may play an important role in the p53 pathway. Similarly, the two tumor necrosis factor superfamily genes TNFRSF6 (Fas/Apo-1) and TNFRSF10B (DR5) along with two genes with zinc finger domains (ZNF148 and ALTE) were tightly clustered together (Fig. 5B). The tumor necrosis factor superfamily genes are involved in p53-dependent induction of apoptosis, and ZNF148 is a transcription factor shown recently to stabilize p53 (Bai and Merchant 2001).
Examples of sets of IR-responsive genes that belong to the same cluster, independent of IR dosage. Dose and cluster membership (see Fig. 4) are indicated to the right of each graph.
We recognize that the likelihood for small groups containing three or four genes to cluster together by chance is high. Therefore, we examined our data to identify larger clusters that are less likely to occur by chance. Within our SOM data, there are four groups of five genes, one group of six genes, and one group of nine genes that exhibit coordinated transcriptional IR response. Examples are shown in Figure 5, C and D.
DISCUSSION
The cellular response to IR consists of an integrated network of protein signaling and transcriptionally regulated pathways. In this study, we focused on the transcriptional changes resulting from IR insult. By using high-density microarrays in conjunction with quantitative RT–PCR, we have identified a set of IR-responsive genes in lymphoblastoid cells, described their temporal expression profiles, and examined effects of IR dose on these temporal patterns of gene expression. We focused on using dose and time parameters to elucidate the complex transcriptional processes that are involved in response to DNA damage resulting from IR exposure.
The main effect of IR on cells is manifested as genotoxic stress resulting from damaged DNA. Part of the cellular response involves stabilization of p53 protein. This increase in p53 protein levels then causes the induction of many genes including ACTA2, CDKN1A, DDB2, FDXR, GADD45A, PIG3, TNFRSF6, and TNFSF10B (Amundson et al. 1999, 2000; Zhao et al. 2000; Kannan et al. 2001; Tusher et al. 2001). All of these genes are shown to be IR-responsive in our study at both 3 Gy and 10 Gy doses. The up-regulation of these genes leads to a diverse set of events, including cell cycle arrest, DNA repair, and apoptosis. Specifically, in the case of TNFRSF6 and TNFRSF10B, these death receptors initiate FADD-dependent activation of caspase activity upon ligand binding. Recent studies have shown that IR can act as a response-enhancing agent for both TNFRSF6 and TNFRSF10B-mediated apoptosis (Reap et al. 1997; Sheikh et al. 1998; Nishioka et al. 1999; Gong and Almasan 2000; Sheard 2001; Embree-Ku et al. 2002). This evidence lends further support for the major role that these death receptor pathways may play in IR-related apoptosis.
Comparison of gene expression profiles for the 3 Gy and 10 Gy IR-responsive genes revealed pertinent characteristics of the pathways involved in response to IR stress. The IR-responsive genes displayed either an early response to IR damage within the first 2 h after IR exposure or a late response that does not become apparent until after 6 h or more following IR treatment.
A number of early IR-responsive genes exhibited marked differences in temporal expression pattern as a result of increased IR dose. Most of these genes displayed transient expression changes that peaked 2 h following 3 Gy of IR exposure and returned to basal levels by 6-h postirradiation. However, at 10 Gy, the same genes responded even faster by showing a rapid increase in gene expression at 1 h postirradiation, and the expression levels remained high throughout the 24-h time course. In addition, a higher radiation dose caused a larger magnitude of change in gene expression, although the increase in magnitude was not proportional to the increase in IR dose. The higher radiation dose generated more DNA damage, which likely caused a more rapid response by the early IR-responsive genes. Functionally, several early IR-responsive genes act as immediate effectors of check-point control and activators of repair and apoptotic pathways. Many of these genes are primary targets of p53, and others may be direct targets of signaling pathways involved in IR damage detection. These genes are crucial in the first steps of properly dealing with IR stress. Therefore, early IR-responsive genes must be quick to react to IR exposure in order to prevent propagation of the detrimental effects of IR.
On the contrary, most late IR-responsive genes exhibited similar temporal gene expression patterns even with varied IR dose. These genes are most likely downstream targets of early IR-responsive effectors. A large number of these late responders were down-regulated gradually during the 24-h time course, which may be due to the shutdown of various pathways as a result of cell death. Other late IR-responsive genes are cell cycle-related or cell cycle-regulated genes. The apparent increase or decrease in expression levels of some of these genes is due to cell cycle arrest and synchronization of the cell population following DNA damage.
We also found sets of genes whose expression profiles are highly correlated at the 3 Gy and 10 Gy doses. Although the pathways regulating the expression profiles of these genes are unknown, their coordinated responses suggest coregulation through common regulatory elements.
Specifically, following 10 Gy of IR treatment, several MAP kinase and MAP kinase-related genes are transcriptionally induced. MAP kinases have been implicated in a variety of biological responses, one of which is stress-induced apoptosis. The MAP kinase pathway is comprised of three distinct components, ERK, JNK, and p38. JNK and p38 are stimulated by various stress and pathogenic insults, whereas ERK responds to mitogenic and differentiation signals (for review, see Herr and Debatin 2001). Although MAP kinases act at the protein level in signaling cascades, the alteration in their mRNA levels may be secondary effects of pathway activation. ATF3 and PPM1D, which exhibit dramatic increases in expression levels following IR stress, may be activated through MAP kinase signaling. ATF3, a member of the mammalian ATF/CREB protein family of transcription factors, is induced by a variety of stress conditions (Chen et al. 1996). Previous reports suggest that ATF3 induction may be due to MAP3K1 (MEKK1) and JNK activation (Liang et al. 1996; Cai et al. 2000). Recent evidence has shown oncogenic properties related to the amplification of PPM1D (Bulavin et al. 2002; Li et al. 2002). PPM1D induction results from p38 activation and mediates negative feedback regulation of p38 MAPK-p53 signaling in response to UV (Fiscella et al. 1997; Takekawa et al. 2000). However, its role in IR stress remains unclear.
Many downstream targets of interferon were induced gradually following IR treatment, even though interferon transcripts did not show detectable changes in transcript levels. It is known that in response to viral infections, interferon operates through the JAK-STAT pathway to mediate transcriptional changes in target genes. This results in antiproliferative effects, which help to suppress viral replication. In the case of IR stress, interferon activity may be promoting the same effects to prevent propagation of DNA damage. In addition, some studies have shown that interferon and other cytokines can sensitize tumor cells to IR damage (Sirota et al. 1996; Gruninger et al. 1999; Schmidberger et al. 1999; McKinney et al. 2000; Nikitina and Gabrilovich 2001).
Specific to the 3 Gy experiments, RNA processing genes including many hnRNPs, and splicing factors were gradually down-regulated during the 24-h time course. There are previous reports of cleavage of some hnRNP proteins as a result of apoptosis, including IR-induced apoptosis (Waterhouse et al. 1996). Others have reported increase in some hnRNPs following UV stress with certain hnRNPs accumulating in the cytoplasm by a p38-dependent pathway (Sheikh et al. 1997; van der Houven van Oordt et al. 2000). However, to our knowledge, evidence for decrease in transcript levels of hnRNPs and splicing factors found in the present study has never been reported.
The findings in this study elucidated parts of the intricate network of genes that are involved in the IR-response. A better understanding of the molecular components and pathways involved in cellular IR stress response will improve our understanding of complex processes such as carcinogenesis and radiation sensitivity. Medically, some of the IR-induced genes can be used as molecular markers for IR exposure.
METHODS
Tissue Culture
Ten lymphoblastoid cell lines (Coriell Cell Repositories) from members of the Centre d'Etude du Polymorphisme Humain (CEPH) families were grown at a density of 5 × 105 cells/mL in RMPI 1620 with 15% FBS. These individuals are not known to be related. Equal numbers of males and females were chosen. Cells were irradiated at 3 Gy and 10 Gy in a 137Cs irradiator. Cells were harvested prior to irradiation (0 h) and at 1, 2, 6, 12, and 24 h after IR exposure. Total RNA was extracted using RNeasy mini-kit (QIAGEN). A reference sample consisting of unirradiated cells from a different set of 10 CEPH individuals was also prepared in the same manner.
Probe Preparation and Hybridization
Total RNA from the experimental and reference groups were pooled according to time point and IR dose. Each pooled total RNA sample (5 μg) was used for probe preparation and hybridization onto U95A GeneChip arrays according to the manufacturer's suggestion (Affymetrix).
Data Analysis
For each gene, expression change was calculated by comparing the signal intensity of the irradiated sample (E) to that of the unirradiated reference sample (B) using d-Chip (Li and Wong 2001). Genes were considered IR-responsive if all of the following conditions were met: (1) E and/or B exceed a threshold, |E-B| > 25th percentile of signal intensities of all the genes assayed; (2) Changes in signal intensity exceed a threshold, the lower confidence bound of the 90% confidence interval for E/B or B/E is >1.5; (3) E and B are significantly different with nominal P < 0.05 (t-test).
Quantitative RT–PCR
We selected 13 genes from the 126 IR-responsive genes that are in common between the two IR doses for validation. Gene expression changes were assayed in lymphoblastoid cells from eight CEPH individuals not used in the U95A GeneChip experiments. Each individual was assessed separately (no pooling of RNA) at the 12-h postirradiation time point following 10 Gy of IR exposure. Pooled total RNA samples (2 μg) were reverse transcribed in a total volume of 100 μL, and diluted to 400 μL. The diluted cDNA (2.5 μL) was used as template for each quantitative PCR reaction. Primers for specific genes were designed using Primer Express software (Applied Biosystems). Quantitative PCR was carried out following the SYBR Green protocol (Applied Biosystems). Assays were performed in triplicate. Relative expression levels were obtained by calculating ddCt normalized to β-actin levels.
Acknowledgments
We thank Drs. Gerd A. Blobel, Greg H. Enders, Warren J. Ewens, Alan M. Gewirtz, Haig H. Kazazian Jr., and Richard S. Spielman for their comments and suggestions. K.Y.J. is supported by NIH training grant GM08216. This work was supported by NIH grant DC00154 and the W.W. Smith Endowed Chair (V.G.C.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
[Supplemental material available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1240103. Article published online before print in August 2003.
-
↵1 Corresponding author. E-MAIL vcheung{at}mail.med.upenn.edu; FAX (215) 590-3709.
-
- Accepted June 9, 2003.
- Received February 3, 2003.
- Cold Spring Harbor Laboratory Press








