
High-Throughput Imaging of Brain Gene Expression
- Vanessa M. Brown1,2,
- Alex Ossadtchi3,
- Arshad H. Khan1,2,
- Simon R. Cherry1,2,4,
- Richard M. Leahy3, and
- Desmond J. Smith1,2,5
- 1Department of Molecular and Medical Pharmacology, 2Crump Institute for Molecular Imaging, School of Medicine, University of California, Los Angeles, California 90095, USA; 3Department of Electrical Engineering, Signal and Image Processing Institute, School of Engineering, University of Southern California, Los Angeles, California 90089, USA
Abstract
Voxelation is a new method for acquisition of three dimensional (3D) gene expression patterns in the brain. It employs high-throughput analysis of spatially registered voxels (cubes) to produce multiple volumetric maps of gene expression analogous to the images reconstructed in biomedical imaging systems. Using microarrays, 24 voxel images of coronal hemisections at the level of the hippocampus of both the normal human brain and Alzheimer's disease brain were acquired for 2000 genes. The analysis revealed a common network of coregulated genes, and allowed identification of putative control regions. In addition, singular value decomposition (SVD), a mathematical method used to provide economical explanations of complex data sets, produced images that distinguished between brain structures, including cortex, caudate, and hippocampus. The results suggest that voxelation will be a useful approach for understanding how the genome constructs the brain.
[All study results are available as a web supplement athttp://www.pharmacology.ucla.edu/smithlab/genome_research_data and at http://www.genome.org.]
Important insights into gene networks in unicellular systems have been obtained using high-throughput multiplex gene expression methodologies, including microarrays (Brown and Botstein 1999), gene chips (Lipshutz et al. 1999), and serial analysis of gene expression (SAGE) (Velculescu et al. 1995). However, these powerful techniques have not yet been applied to understanding how the genome constructs the three dimensional (3D) structure of multicellular organisms. In contrast, tools exist for 3D imaging of gene expression in the living organism, but at present these methods only permit the examination of one, or at most, a few, genes at a time (Gambhir et al. 1999; Herschman et al. 2000; Louie et al. 2000; Zacharias et al. 2000). Here, a method called voxelation is described, which uses high-throughput gene expression analysis to produce volumetric expression maps for thousands of genes in parallel. The method gets its name from the term voxel, which is used in biomedical imaging to refer to a 3D image volume element. Voxelation is conceptually simple, and entails the direct creation of voxels (cubes) in spatial register with the brain, together with the application of high-throughput gene expression analytic techniques to RNA extracted from the voxels. The resulting maps of gene expression are analogous to the images reconstructed in biomedical imaging systems, such as CT and PET.
RESULTS
Coronal hemisections at the level of the hippocampus of a normal human brain and an Alzheimer's disease brain were divided into 24 voxels (Fig. 1A) and analyzed using 2000 gene microarrays. To provide an overall survey of the data, gene expression correlation matrices for both specimens were constructed (Fig. 1B). The genes in the normal matrix were parsimoniously clustered based on minimization of a cost function related to K-means, resulting in a cluster number of five. The same gene order was used to construct the corresponding matrix for the Alzheimer's hemisection. Strikingly, the matrices for both specimens were very similar as judged using a Monte-Carlo simulation (P < 0.0001), demonstrating excellent reproducibility of the voxelation strategy. To gain further insights into gene expression in healthy and diseased brain, a subset of the data was extracted. This subset consisted of the genes in common between both the normal and Alzheimer's hemisections, where the genes had a spatial expression correlation coefficient of >0.92 with at least one other gene in the same brain. This procedure should identify networks of coregulated genes in both brains. Gene expression correlation matrices for the coregulated subsets were created (Fig. 1C; Table 1), with the normal matrix ordered using a similarity metric, and the Alzheimer's matrix following suit. Similar to what was seen for the overall data, there was a striking correspondence between the two matrices for the normal and Alzheimer's hemisections. Again, this concordance was highly significant, as judged using a Monte-Carlo simulation (P < 0.0001), implying that the coregulated networks of genes are independently maintained in both the normal and Alzheimer's specimens.
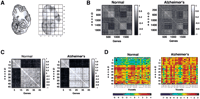
Correlated gene clusters. (A) Representation of the voxelation process on a normal hemisection. Abbreviations: Ca, tail of caudate nucleus; Cx, cortex; Hi, hippocampus; Pu/GP, putamen/globus pallidus; Th, thalamus. (B) Gene expression correlation matrices for the normal and Alzheimer's hemisections. The correlation of expression levels across voxels between any two genes is read by looking along the relevant row and column, and finding the intersection. The darkness of the corresponding element gives the correlation between that pair of genes by reference to the scales (right). The diagonals are the autocorrelations of the gene expression patterns for each gene and are (and should be) equal to one. All other correlations must be between 1 and −1. The genes are parsimoniously ordered in the normal correlation matrix, giving five clusters. The order of genes in the Alzheimer's matrix follows the normal. (C) Gene expression correlation matrices for the subset of genes common to both specimens that display a spatial expression correlation coefficient of >0.92 with at least one other gene within the same brain. The genes in the normal correlation matrix are ordered using a similarity metric, and the order of genes in the Alzheimer's matrix is the same as for the normal. Two mutually exclusive clusters of coregulated genes are present: cluster 1 (genes 1–14) and cluster 2 (genes 15–46). In both (B) and (C), the similarity of the correlation matrices between the two specimens is highly significant, as judged using a Monte-Carlo simulation. (D) Spatial gene expression patterns for the subset of correlated genes. The voxels are laid out in linear fashion forming the columns of the matrices, whereas the genes form the rows. The relative level of expression of a gene in any particular voxel can be deduced by reference to the scales below. The two clusters of genes are apparent, and although each cluster consists of highly correlated expression patterns within both the normal and Alzheimer's hemisections, the patterns of gene expression are different between the two hemisections.
Co-Regulated Genes
To further examine replicability between, as well as within, the hemisections, the voxels were placed in ascending order (A2, B1, B2, . . . . .), with the first member of the series (A2) being counted as 1 (i.e., odd), the second (B1) as 2 (i.e., even), etc. The data presented in Figure 1C was then arbitrarily split into two parts for each hemisection, consisting of even and odd numbered voxels. Based on the Monte-Carlo strategy, there was highly significant similarity among the data sets (odd and even voxels), both between and within hemisections (P < 0.0001), further demonstrating the reproducibility of voxelation (data not shown).
Interestingly, the correlation matrices of the coregulated subset shown in Figure 1C revealed two mutually exclusive clusters. Cluster 1 (genes 1–14) was positively correlated within itself, and negatively correlated with cluster 2 (genes 15–46), and vice versa. The spatial map of gene expression variation across the voxels for the selected subset of genes in both specimens is shown in Figure 1D. The figure demonstrates that although the mutually dependent network of spatially coregulated gene clusters is maintained within each brain, the expression patterns are different in the Alzheimer's specimen compared to the normal, particularly for cluster 1. There were some interesting biological relationships within the coregulated subset of genes. U5-100K (gene 4, cluster 1) andRNPS1 (gene 16, cluster 2), have highly negatively correlated spatial expression patterns in both the normal and Alzheimer's hemisections, as indicated by their membership in the two separate clusters. Both these genes encode proteins with similar functions, U5-100kD being a U5 snRNA associated RNA helicase (Laggerbauer et al. 1998; Teigelkamp et al. 1997), and RNPS1an RNA-binding protein involved in alternative splicing (Loyer et al. 1998; Mayeda et al. 1999). The connected functions of these genes may account for their negatively related spatial expression patterns. A bioinformatics analysis found shared regulatory regions between these genes (below). Another gene, MADD (gene 38, cluster 2), showed elevated expression in the hippocampus of the Alzheimer's hemisection (voxels F2, G1, G2) compared to normal, and this gene is induced in the hippocampus of hypoxic brains (Zhang et al. 1998).
To find control regions shared between the correlated and anticorrelated genes of the subsets shown in Figure 1C,D, a bioinformatics analysis was performed to look for conserved noncoding sequences (Table 2; Fig.2). Gene pairs were analyzed with gene expression correlation coefficients >0.8 or <−0.6.BLAST was used to find homologies, but not provide reliable estimates of their statistical significance, as the algorithm employs asymptotic statistical approximations, which are not accurate for shorter sequences (Benson et al. 2000). The resulting homology regions were further scrutinized for transcription factor binding sites using the TRANSFAC database (Wingender et al. 2000). The homology search was confined to sequences 20-kb upstream, 20-kb downstream, and in all introns of the relevant genes. The analysis revealed a complex array of potential control elements shared between genes, which may be responsible for their expression pattern relationships. Some of the genes (5/9) had putative control regions in the flanking or intron sequences of adjacent genes. In most of these cases (4/5), orthologs of the coregulated gene were found in theDrosophila genome, and in all cases where aDrosophila ortholog existed (4/4), analogous control regions were also found. However, in the Drosophila genome, the putative regulatory regions were found in a distinct context: either in the flanking region or intron of a completely different neighboring gene. This validated the likely relevance of the regulatory region in the original gene of interest. In all cases, except for one (RNPS1 and U5-100K, homology block 2, ggaaggatggt(g/a)tctcctg, respectively), the potential regulatory sequences harbored known transcription factor binding sites. We predict that the one exception may in the future be found to represent an as yet uncharacterized binding site. Nevertheless, the significance of the potential regulatory sequences must be confirmed experimentally.
Potential Regulatory Sequences in Co-Regulated Genes of Normal and Alzheimer's Hemisections
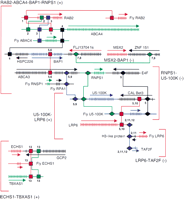
Putative regulatory elements shared between groups of correlated and anticorrelated genes. There were three groups of correlated (+) genes: (1) RAB2, ABCA4, BAP1, RNPS1, (2) U5-100K, LRP6, (3) ECHS1, TBXAS1; and three groups of anticorrelated (−) genes: (1) BAP1, MSX2 (2) RNPS1, U5-100K (3) LRP6, TAF2F. The groups are indicated by square brackets. The regulatory sequences responsible for correlated expression are shown as squares, those responsible for anticorrelated expression are shown as diamonds. Genes are indicated by UniGene symbol or name (http://www.ncbi.nlm.nih.gov/UniGene). Exons are indicated by short vertical lines. Lines delineate the relationships between the conserved regulatory sequences. Multiple control regions frequently connected the genes. Sometimes these control regions were found in introns or flanking regions of adjacent genes. In that case, where there was a Drosophila ortholog of the relevant gene, the control region was conserved in the Drosophila genome but in a different context. Potential binding sites are: (1) OCT-1, (2) HFH8, (3) TFIID, (4) AP1, (5) IK2, (6) Sp1, (7) USF, (8) MYOD, (9) GKLF, (10) IK1, (11) HFH3, (12) XFD1, and (13) AP4.
In addition to global analyses of spatial gene expression in the normal and Alzheimer's hemisections, significant (P < 10−7) gene expression differences when averaged across the voxels were sought between the two specimens (Fig.3A). To assess the replicability of the findings, equivalent voxels (voxel F1) from the hippocampus of an additional normal and an additional Alzheimer's specimen were also analyzed, using a 5000 gene microarray with substantial overlap with the 2000 gene microarray. The F1 voxel was chosen for replication as it is part of the hippocampus, which is strongly affected in Alzheimer's disease. A scatterplot was constructed that compared the expression level differences between normal and diseased specimens using those genes judged significantly different across the entire hemisections and also present on the 5000 gene microarray (Fig. 3B). Despite the fact that the whole hemisections and the F1 voxels came from four entirely different individuals, the scatterplot analysis showed excellent replicability of gene expression differences (P = 0.0002) between the normal and Alzheimer's disease groups. This data suggests that the uncovered differences between the normal and Alzheimer's disease brains represent real distinctions attributable to the disease process, and are not because of the inevitable lack of precisely matched human samples.
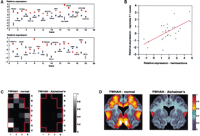
Genes whose average expression across voxels is significantly different in the normal compared to the Alzheimer's brain. (A) Graph showing mean expression levels across the 24 voxels in the normal and Alzheimer's hemisections on a logarithmic scale (log2) (±SEM). Normal: red, Alzheimer's: blue. The genes are ranked from most (gene 1) to least significant (gene 36, P< 10−7). Two genes of the differentially expressed subset, PTPRN2 (genes 2 and 3, upper row) and WASF1 (genes 2 and 3,lower row), were present as duplicate spots on the microarrays, and give an independent assessment of within array replicability. (B) Scatterplot comparing the mean expression differences between the normal and Alzheimer's disease brains based on the hemisection data and the replicate F1 voxel data. Expression differences are shown using the logarithm (log2) of the gene expression ratios between the normal and diseased specimens. The genes employed in the scatterplot are those judged significantly (P< 10−7) different when averaged across the whole hemisections and which are also present on the 5000 gene microarray used to analyze the replicate F1 voxels. A total of 27 genes resulted (YWHAH, PTPRN2, ARL6IP, ICAP-1A, DRAP1, SMS, SEPW1, NFATC3, PSCD2, XPO1, ZNF142, PALLADIN, RAP2A, BICD1, LOC51628, DSCR1L1, WASF1, RARS, CCS, TIF1α, PRKCB1, SALL2, MAPK10, IDH3A, IDI1, TAF2F, DNCI1). There was a highly significant correlation between the data from the hemisections and the F1 voxels (r = 0.65, F[1,25] = 18.34, P = 0.0002). The best fit using least squares linear regression is shown. (C) An example of the spatial expression pattern of a gene (YWHAH) whose expression is significantly greater in the normal compared to the Alzheimer's brain. The level of gene expression can be deduced by reference to the scale on the right. (D) YWHAH expression patterns after smoothing over voxels using imaging software, and projecting onto the relevant neuroanatomy. The resulting images were reflected along the midline for the figure, giving bilateral symmetry.
A number of intriguing genes were found to be significantly different between the normal and Alzheimer's disease hemisections (Fig. 3A; Table 3), involved in such diverse areas as signal transduction (e.g., YWHAH, PTPRN2,RAP2A), modulation of the cytoskeleton (e.g., ICAP-1A,PALLADIN), transcription (e.g., DRAP1, TIF1α, NFATC3, TAF2F), and cholesterol synthesis (IDI1). There were also two novel genes. Interestingly, it has been reported that the expression within hippocampus and neocortex of one of the differentially expressed genes, MAPK10, closely matches that of Alzheimer disease targeted neurons (Mohit et al. 1995). The vast majority of the genes are expressed more highly in the normal brain than the Alzheimer's brain (29/34). This is a highly significant deviation from random (χ2 = 18.74, df = 1, P< 0.0001), and possibly reflects the considerable neuronal cell death that occurs in Alzheimer's disease.
Genes Differentially Expressed in Normal and Alzheimer's Hemisections
A graphic presentation of the spatial expression pattern across voxels for one of the significantly differentially expressed genes,YWHAH, is shown in Figure 3C for both the normal and Alzheimer's hemisections. In Figure 3D, a Bayesian approach to creation of expression images for YWHAH was employed, using a prior assumption of nearest neighbor continuity. This resulted in smoothed expression patterns over the voxels, which were then projected onto the relevant neuroanatomy and reflected along the midline, giving bilateral symmetry.
Singular value decomposition (SVD) is a powerful method for economical descriptions of complex data sets (Hendler and Shrager 1994; Frackowiak et al. 1997; Alter et al. 2000). This statistical method reduces dimensionality, while retaining the maximum possible fraction of the variance from the original data. For example, when used in biomedical imaging, SVD analysis frequently explains data sets on the basis of known functional and anatomical boundaries (e.g., cortical vs. subcortical). In the context of gene expression patterns, it might be expected that SVD would show which sets of genes (“vectors”) account for the major variations between the voxels, and hence which sets of genes play important roles in setting up spatial patterns of differentiation in the brain. In essence, the gene vectors would represent ‘votes‘ for the properties of the various brain regions in which they are manifest. It should be noted that SVD does not rely on preconceived notions or hypotheses, and is entirely data driven. To see if SVD would illuminate the large amounts of data from the voxelation studies of the normal and Alzheimer’s hemisections, we performed an analysis on the conjoint matrix resulting from the top 120 genes most strongly differentially expressed between the samples (P ∼ 0.05) (c.f. Fig. 3). The results of the SVD analysis are presented in Figure 4. The first principal component (PC) was uniformly expressed, and represents genes consistently differentially expressed across all voxels. Analogously, the first PC in biomedical imaging studies is often an average representation of the entire brain. The second PC is largely restricted to cortex, the third to both the tail of the caudate and the hippocampus, and the fourth to the insular cortex. This restriction to anatomical regions is remarkable considering the two-fold uncertainty in the microarray data, the relatively crude spatial maps (24 voxels), and the inevitability, given the nature of human samples, that the two hemisections are not perfect controls for each other. With increased resolution and more comprehensive gene surveys, voxelation may ultimately reveal the molecular ontology of the brain, demonstrating which parts of the brain are most closely related in terms of gene expression patterns to other parts.
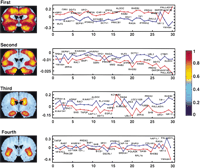
SVD delineates anatomical regions of the brain. The conjoint matrix resulting from the top 120 genes most strongly (P ∼ 0.05) differentially expressed between the normal and Alzheimer's hemisections was analyzed. The spatial patterns resulting from the first, second, third, and fourth PCs are shown. Alongside are the first 30 members of the corresponding gene vectors. The ordinate represents the contribution by the relevant gene to the variation of the vector spatial pattern, whereas the abscissa represents the genes in decreasing order of significance of differential expression. The genes are indicated by UniGene symbol or name. Normal: red, Alzheimer's: blue. The first component is uniformly expressed over the brain, and represents an image of average gene expression differences between the samples. The second component is largely restricted to cortex, the third to both the tail of the caudate and the hippocampus, and the fourth to the insular cortex. The level of expression of the relevant gene vector in the spatial patterns can be deduced by reference to the pseudocolor scale (right). Imaging software smoothed the expression patterns over the voxels, and the hemisection was reflected along the midline for the figure, giving bilateral symmetry.
DISCUSSION
The investigations reported here demonstrate that employing spatial information from whole organisms together with high-throughput gene expression methodologies will provide valuable additional insights not easily obtained from studies of unicellular systems. Although the voxelation studies had limited spatial resolution, useful data was obtained, and there are parallels with functional imaging of the brain, which gives important insights despite the fact that the voxels are inhomogeneous (Raichle 1998). The spatial information content of voxelation helped define control regions in networks of coregulated genes, and further insights were obtained from SVD. It should be emphasized that these conclusions do not depend on the assumption of precisely matched samples. For example, the networks of coregulated genes were clearly conserved between the two hemisections across multiple voxels, despite the inevitable lack of exact controls using human specimens. This lack notwithstanding, consistent gene expression differences between normal and Alzheimer's disease brains were found.
Despite the drawbacks of human studies, by definition these investigations have the advantage of disease validity. In contrast, studies using mice can be precisely and accurately controlled, and furthermore provide opportunities for the use of genetically engineered animals. However, with mice there will always be unresolved uncertainties over disease model validity (especially where the etiology is unclear, e.g., the neuropsychiatric disorders such as schizophrenia). In the longer term, perhaps the most information can be extracted by the judicious combined use of both humans and mice, as well as other model systems. A relevant point here is that the same volumetric resolution (voxel size), will yield better relative resolution with larger brains. For example, identical voxel dimensions will produce about a seven-fold higher relative resolution using the rat brain compared to the mouse, because of the corresponding brain volumes of these species.
An important future task for voxelation will be to increase the amount of information it provides, by miniaturization of voxel size to improve resolution and also analysis of increased numbers of genes. The direct incorporation methodology for probe labeling employed in this study is sufficiently sensitive to allow construction of 13,000 voxel maps of the human brain. In principle, more sensitive techniques, such as those using tyramide signal amplification, should allow construction of 325,000 voxel images. By comparison, a modern CT or PET scan of the human brain typically employs about 150,000 voxels. Because of the much smaller size of the mouse brain, it is not feasible to use direct incorporation for construction of spatial expression maps of single brains in this organism. However, pooling spatially equivalent voxels will allow decreased voxel size, and hence improved resolution, while still allowing recovery of sufficient RNA for analysis. For individual mouse brains, tyramide signal amplification will permit construction of 75 voxel maps. Real-time quantitative RT–PCR is still more sensitive, and will allow construction of 6000 voxel maps, although automation and miniaturization will doubtless be required to harvest such small voxels. Real-time quantitative RT–PCR has lower throughput than microarrays, but the potential of PCR for automatability and scalability will nevertheless allow such methods in combination with voxelation to surpass the throughput of classical techniques, such as in situ hybridization.
It will also be important to find ways to drive down costs. Although microarrays are a relatively cheap tool on a per gene basis, voxelation will become increasingly expensive as greater numbers of voxels are analyzed in the quest for improved resolution in a variety of experimental situations. Furthermore, as resolution is pushed ever higher, computational analysis will become an important issue because of the overwhelming amounts of data. However, assuming Moore's law continues to hold true, improvements in computing power should allow data analysis to keep pace.
All of these goals—higher resolution, better analytic methodologies, higher throughput and more powerful computational tools—will provide substantial challenges. Ultimately, however, cross-species high-resolution voxelation of healthy and diseased brains is likely to provide better comprehension of the logic of the genome, and how this program goes awry in disorders affecting the brain. Such investigations will give important information on the genomic construction of the brain as well as novel starting points for therapy.
METHODS
Voxelation Procedure
The hemisections from both the normal and Alzheimer's brain were 8 mm thick, and were from the left side at the level of the hippocampus, corresponding to section 17 of the University of Maryland Brain and Tissue Bank protocol, method 2 (Brain and Tissue Bank, University of Maryland, http://medschool.umaryland.edu/BTBank). In each case, the voxelation was performed using a 32-voxel template consisting of eight rows in the superior/inferior axis (A to H, superior to inferior), and four columns in the medial to lateral axis (1 to 4, medial to lateral). The two hemisections were of different superior/inferior and medial/lateral dimensions, and therefore the voxelation template of the Alzheimer's brain was linearly spatially deformed along these axes relative to the normal brain, so that the same number of potential voxels were present in both templates. Subsequent computational adjustment, based on the anatomical topography of the two hemisections, allowed for complete gene expression image registration. Because the brain hemisections were roughly semicircular, whereas the voxelation template was rectangular, some voxels in the templates were empty. A scheme was established a priori to deal with voxels on the edge of the brain, whereby if the volume of biological material in the voxel was <50% voxel volume, those voxels were pooled with adjacent voxels. The following clockwise scheme was employed to pool voxels until a combination >50% was possible: First the subthreshold voxel was combined with the voxel medially, then superiorly, then laterally, then inferiorly. If an edge voxel contained more biological material than 50% of the voxel volume, it was considered a free-standing image element. The scheme resulted in the following 24 data voxels in common for the two hemisections: A2, B1, B2, B3, C1, C2, C3, D1, D2, D3, D4, E1, E2, E3, E4, F1, F2, F3, F4, G1, G2, G3, H2, H3. The voxel grid is shown in Figure 1A. The normal brain was from a 49 yr old male who died as a result of a car accident. The postmortem interval was 9 h. The Alzheimer's brain was Lewy body positive, and was from an 85 yr old female who died from cardiac complications. This individual had dementia with accompanying depression and delusions, and was taking sertraline and haloperidol. The postmortem interval was 12 h. The normal F1 voxel was from a 22 yr old male who died as a result of atherosclerotic cardiovascular disease. The postmortem interval was 4 h. The Alzheimer's disease F1 voxel was from an 85 yr old female, with well-formed neuritic plaques and scattered neurofibrillary tangles. The case was classified as high likelihood of Alzheimer's disease based on consensus recommendations (National Institute on Aging 1997). The cause of death was respiratory failure and the postmortem interval 10 h.
Microarray Analysis
For each voxel of the normal and Alzheimer's hemisections, 100 μg of Cy3-labeled voxel RNA and 100 μg of Cy5-labeled control RNA were cohybridized to a separate 2000 gene microarray, as described previously (Eisen and Brown 1999). The control RNA was used to facilitate interarray comparisons, and consisted of total RNA from the normal hemisection reconstructed by combining proportionate amounts of RNA from each voxel. For each gene, signal to noise ratio was 2.5-fold above background for both the Cy3 and Cy5 channels. For the F1 voxels, two experiments were performed in which labeled normal and Alzheimer's RNA were directly compared by cohybridization to separate 5000 gene microarrays, but with the Cy3 and Cy5 dyes reversed for the second experiment. Gene expression values were taken as the mean of the two experiments. Of the genes present on the 2000 gene microarray, 62% were also present on the 5000 gene microarray.
The microarray data was processed using two types of normalization procedures. First, spatial trends existing in the data attributable to chip printing were removed by nonlinear transformation of the data sets. The second normalization procedure was designed to compensate for differences in the labeling and chemical properties of the Cy3 and Cy5 dyes, by aligning the histograms of the dye signals both within, as well as between, chips. The genes chosen for the microarrays were a random selection of sequence verified known and novel cDNAs obtained from Research Genetics. The genes are listed on the study web site (below).
Correlation Matrix Clustering
The genes in the omnibus normal correlation matrix of Figure 1B were clustered using an algorithm related to the K-means procedure (Sherlock 2000). The algorithm was based on minimization of a cost function, C(K) = Σ(distribution within clusters)2 + K2, where K is the number of clusters. As the number of clusters goes up, the first term of the equation decreases, whereas the second increases, and the C(K) is hence expected to show a minimum. The genes in the Alzheimer's correlation matrix were placed in the same order as the normal. For the correlated subset matrices shown in Figure 1C, the genes in the normal matrix were ordered using a hierarchical clustering approach with a similarity metric related to the centroid method (Milligan 1980). The first row of the matrix was chosen to exhibit a strong contrast between the highest and lowest correlation coefficient for that row. This row was denoted as the base vector, B, with respect to which the remaining rows, R, were arranged in order of decreasing similarity, using a metric consisting of Σi(Bi − Ri)2, where i = the elements of the rows. Once the matrix for the normal brain was created, the matrix for the Alzheimer's brain was created following the same order.
Monte-Carlo Simulations
The Monte-Carlo simulation to assess the similarity of the normal and Alzheimer's correlation matrices in Figure 1B employed random permutation of the columns of the matrices, and showed that the similarity was highly significant (P < 0.0001). For the simulation, the discrepancy between randomly selected pairs of permuted matrices was quantitated using the Frobenius norm of the matrix obtained by subtracting one permuted matrix from the other. The difference between the mean of the resulting distribution and the Frobenius norm obtained from the actual normal and Alzheimer's matrices was used to show significance. The Monte-Carlo simulation to assess the similarity of the normal and Alzheimer's correlation matrices in Figure 1C also showed high significance. The simulation employed random substitution of genes drawn from the entire 2000 gene dataset in the rows and columns of the matrices. Significance was assessed using Frobenius norms, as described above.
Singular Value Decomposition
The conjoint matrix employed for SVD was obtained using the top 120 genes most strongly differentially expressed between the normal and Alzheimer's hemisections (P ∼ 0.05). The matrices ofm voxels × n genes for the normal and Alzheimer's specimens were concatenated along the spatial dimension, giving a matrix of size m × 2n. The concatenation procedure provided a common spatial dimension for the data sets of both samples. When the number of genes in the SVD analysis was limited to the 34 most significant (P < 10−7) differentially expressed genes (Fig. 3) rather than the top 120, the spatial expression patterns of the first and second PCs were preserved, whereas the patterns of the third and fourth were altered. This observation implies superior robustness of the first and second PCs, and it is typical of SVD that the first few PCs account for much of the data.
Web Sites
All study results are available as a web supplement athttp://www.pharmacology.ucla.edu/smithlab/genome_research_data andhttp://www.genome.org.
WEB SITE REFERENCES
http://medschool.umaryland.edu/BTBank, Brain and Tissue Bank, University of Maryland.
http://www.ncbi.nlm.nih.gov/UniGene, UniGene web site.
Acknowledgments
This research was supported by grants from the Dana Foundation, Merck Genome Research Institute, Staglin Music Festival and NARSAD Young Investigator Award, W.M. Keck Foundation, National Foundation for Functional Brain Imaging, NIH, NSF, and UCLA School of Medicine. Specimens were obtained from the University of Maryland Brain and Tissue Bank under NIH contract N01-HD-1-3138, and the National Neurological Research Specimen Bank, VAMC, Los Angeles, sponsored by NINDS/NIMH, National Multiple Sclerosis Society, VA Greater Los Angeles Healthcare System, and Veterans Health Services and Research Administration.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Department of Biomedical Engineering, One Shields Ave, University of California, Davis, CA 95616, USA.
-
↵5 Corresponding author.
-
E-MAIL DSmith{at}mednet.ucla.edu; FAX (310) 825-6267.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.204102.
-
- Received July 6, 2001.
- Accepted October 26, 2001.
- Cold Spring Harbor Laboratory Press








