
Sequence-Based Design of Single-Copy Genomic DNA Probes for Fluorescence In Situ Hybridization
Abstract
Chromosomal rearrangements are frequently monitored by fluorescence in situ hybridization (FISH) using large, recombinant DNA probes consisting of contiguous genomic intervals that are often distant from disease loci. We developed smaller, targeted, single-copy probes directly from the human genome sequence. These single-copy FISH (scFISH) probes were designed by computational sequence analysis of ∼100-kb genomic sequences. ScFISH probes are produced by long PCR, then purified, labeled, and hybridized individually or in combination to human chromosomes. Preannealing or blocking with unlabeled, repetitive DNA is unnecessary, as scFISH probes lack repetitive DNA sequences. The hybridization results are analogous to conventional FISH, except that shorter probes can be readily visualized. Combinations of probes from the same region gave single hybridization signals on metaphase chromosomes. ScFISH probes are produced directly from genomic DNA, and thus more quickly than by recombinant DNA techniques. We developed single-copy probes for three chromosomal regions—the CDC2L1 (chromosome 1p36), MAGEL2(chromosome 15q11.2), and HIRA (chromosome 22q11.2) genes—and show their utility for FISH. The smallest probe tested was 2290 bp in length. To assess the potential utility of scFISH for high-resolution analysis, we determined chromosomal distributions of such probes. Single-copy intervals of this length or greater are separated by an average of 29.2 and 22.3 kb on chromosomes 21 and 22, respectively. This indicates that abnormalities seen on metaphase chromosomes could be characterized with scFISH probes at a resolution greater than previously possible.
Conventional fluorescence in situ hybridization (FISH) commonly uses cloned genomic probes for hybridization to fixed, denatured chromosomes. These genomic probes are generally large and most often cloned into vectors, such as cosmids, yeast, or bacterial artificial chromosomes that accept 50 kb to megabase-sized genomic inserts (Trask et al. 1993; Bray-Ward et al. 1996; Korenberg et al. 1999). Because these probes contain both single-copy and repetitive DNA sequences, specificity for the single-copy chromosomal sequences is achieved by enriching for single-copy sequences (Fuscoe et al. 1989) or by disabling hybridization of the repetitive components. Blocking of repetitive sequence hybridization is typically performed by preannealing the probe with an excess of unlabeled repetitive DNA prior to duplex formation with chromosome specific targets (Sealey et al. 1985; Lichter et al. 1988; Pinkel et al. 1988). Single-copy sequence enrichment has also been achieved by removing repetitive sequences with column purification (Craig et al. 1997).
Repetitive sequences comprise almost 50% of the human genome (Britten and Kohne 1965; Britten and Davidson 1976). There are at least 480 identified repetitive sequence families, and they are diverse in frequency and sequence heterogeneity (Jurka 1998). These repetitive DNA sequences, expressed genes, and single-copy sequences can be located precisely in draft and complete genomic sequence contigs by computational methods. We describe a method to design and produce custom genomic probes from computationally defined, single-copy genomic sequences. Probe sequences are inferred from DNA sequences of larger genomic intervals of interest with software that determines the locations of repetitive DNA elements contained in these sequences. By excluding the repetitive sequences, probes are designed from 2-kb to 10-kb single-copy (sc) intervals, synthesized in vitro, purified, and detected by FISH to chromosomes (scFISH). We developed scFISH probes from several chromosomal regions. This approach streamlines the development and production of single-copy, sequence-specific hybridization probes for detection of genetic rearrangements in both rare and common chromosome anomalies.
RESULTS
Chromosomal Regions Selected for Probe Design
Single-copy probes were developed for three different autosomal regions according to the scheme shown in Figure1. Probes were produced for sequences within chromosome 1p36, chromosome 15q11.2, and chromosome 22q11.2. Hemizygous deletions of these sequences result in monosomy 1p36, Prader-Willi or Angelman, and DiGeorge syndromes, respectively. Genomic sequences ∼100 kb in length corresponding to the CDC2L1,MAGEL2, and HIRA cDNAs were identified by analysis (Altschul et al. 1990) of the human draft and complete sequence contigs (Table 1). The locations of the longest single-copy intervals were then deduced by sequence analysis of the HIRA (22q11.2; GenBank accession no. NT_001039), MAGEL2 (also denoted NDNL1; 15q11.2; accession no. AC006596), and CDC2L1 (1p36; accession no. AL031282) genomic sequences. The HIRA genomic sequence contained 10 single-copy segments ≥2 kb in length, of which the four longest were selected for probe design. The HIRA products were 5170, 3691, 3344, and 2848 bp in length (Fig. 2, lanes 3–6); separated by an average of 14.3 kb, and contained within a single 58.1-kb interval. Multiple single-copy intervals ≥2 kb were also identified in the genomic sequences containing MAGEL2and CDC2L1. The longest clusters of single-copy intervals were selected to produce a discrete hybridization pattern on metaphase chromosomes. The probes from the MAGEL2 locus (4100 bp, 3544 bp [Fig. 2, lane 2] and 2290 bp [Fig. 2, lane 1] in length) were all contained within a 26.5-kb interval, with a 15.6-kb gap separating the 2290-bp and 4100-bp segments. The CDC2L1 probes consisted of two fragments, 4823 bp and 4724 bp (Fig. 2, lanes 7,8), together comprising a contiguous 9.6-kb single-copy sequence. A 2378-bp product, which represented a portion of the 4823-bp probe (positions 9137–11515 of AL031282), was also generated for comparative hybridization studies.

The process for developing and producing scFISH probes. (A) Computational sequence analysis of genomic sequence. Genomic sequence is compared with a database of repetitive sequences to identify the positions of repeat elements (left arrows or right arrows), thus delineating the boundaries of single-copy intervals (solid lines). These deduced sequences are then compared with the genome sequence to detect potential gene family members, pseudogenes, and other complex low-copy repeats (data not shown). The intervals are then sorted according to their respective lengths, and primers (⇀ and ↽) are optimized to amplify intervals with lengths that exceed a threshold (i.e., 2 kb). (B) Single-copy intervals are amplified by long PCR of high molecular weight genomic DNA. The PCR products are then purified, labeled by nick translation (underlined asterisks), hybridized, detected with a fluorescent antibody that recognizes the labeled nucleotide, and visualized by fluorescence microscopy.
Definition of scFISH Probes
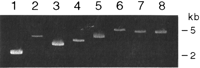
Long PCR amplification. Amplification products are separated by gel electrophoresis in 1% low-melt temperature agarose in modified TAE (40 mM Tris-acetate, pH 8.0, 0.1 mM Na2EDTA). Lanes 1and 2 show the 2290-bp and 3544-bp products from theMAGEL2 locus; lanes 3– 6 contain, respectively, the 2848-bp, 3344-bp, 3691-bp, and 5170-bp products from the HIRA locus; lanes 7 and 8 are the 4724-bp and 4823-bp products, respectively, from the CDC2L1 locus.
Sequence Analysis of scFISH Probes
We analyzed the sequences of scFISH probes to determine how the constraints of probe design (i.e., the absence of repetitive sequences and suitability for amplification by long PCR) biased the characteristics of the resultant probes. The nucleotide compositions of the probe segments ranged from 61% to 65% cytosine and guanine (% C/G) for the CDC2L1 locus, 41% to 49% forMAGEL2, and 47% to 53% for HIRA. The % C/G of each probe was similar or greater than that of the genomic sequence from which it was derived (54% for CDC2L1, 41% forMAGEL2, and 46% for HIRA) and, in all cases, exceeded the average % C/G of the genome (41%; Gardiner 1995).
Most probes contained both exons and introns, and occasionally multiple exons and introns. The 5170-bp product from the HIRA locus contained exon 13 and introns 12 and 13; the 3691-bp product spanned an interval containing introns 21–24; the 3344-bp product spanned introns 13–15; and the 2848-bp product spanned introns 2–4 (Fig.3A). The 3544-bp MAGEL2 probe began 204 bp downstream from the first coding nucleotide and contained 1789 bp of expressed coding and 3′ UTR sequences as well as 1755 bp of nontranscribed sequence (Fig. 3B). The 4100- and 2290-bpMAGEL2 probes began 20.5 kb and 2.7 kb, respectively, downstream from the MAGEL2 gene (Fig. 3B), and neither sequence is expressed (based on comparisons with dbEST and GenBank). The two probes from the CDC2L1 region were contiguous, beginning within intron 11 and ending within the 3′ untranslated region of the gene (Fig. 3C).
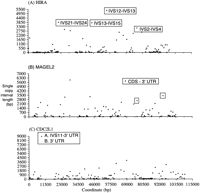
Distribution of single-copy intervals (▪) along genomic sequences containing (A) HIRA, (B)MAGEL2, and (C) CDC2L1 loci. Genomic sequence coordinates are derived from the GenBank accession numbers for each locus (see Methods and Results). Each point corresponds to the length of each single-copy interval on the y-axis at the genomic coordinate. The first position in panel A corresponds to position 798334 of GenBank accession no. NT_001039. TheHIRA gene is transcribed from the antisense strand of NT_001039 and corresponds to positions 900363–799527. The intervals containing probe sequences used in this study are boxed and labeled with the relevant features of genes from these regions. IVS indicates intron; 3′UTR, 3′ untranslated region; and CDS, coding sequence.
We compared probe sequences with available draft or final genome sequences by BLAST analysis. Assuming 91% genomic coverage (http://www.ncbi.nlm.nih.gov/genome/seq/HsHome.shtml, 11/12/00), all of the probe sequences were represented only once in the genome. A subinterval in the 4724-bp CDC2L1 probe showed limited similarity to draft sequences at a second chromosome 1 locus (89% identity over 130 bp) and chromosomes 10 (84% identity over 440 bp) and 15 (84% identity over 516 bp). Other probe sequences showed minimal sequence similarity with nonallelic genomic sequences (≤54 bp). The relatively short lengths of similarity with nonallelic loci and the high stringency of posthybridization wash conditions prevented the detection of chromosomal loci, other than those for which they were designed.
Probe Purification
PCR amplification products from the HIRAand MAGEL2 loci were initially isolated by phenol/chloroform extraction and isopropanol precipitation of the amplification reactions. This approach was unsatisfactory, as the amplification reactions also contained single-stranded PCR extension products with flanking repetitive sequences. These products were simultaneously isolated during the initial extraction, and they nonspecifically hybridized to all chromosomes when preannealing with Cot1 DNA was not performed (data not shown). To remove these extension products and thus prevent nonspecific chromosomal hybridization, the amplicons were size-selected by gel electrophoresis and eluted by spin-column chromatography.
Chromosomal Hybridization Results
Comparative chromosomal hybridizations with and without preannealing of scFISH probes to Cot1 DNA showed that sequence-based selection of probes eliminated repetitive genomic sequences. The purified 3544-bp probe from the MAGEL2 locus, preannealed with Cot1 DNA, hybridized specifically to homologous sequences on chromosome 15 from cells of a normal individual (Fig.4A). The same fragment from theMAGEL2 locus without Cot1 DNA preannealing is presented in Figure 4B. Hybridizations were observed in greater than 90% of metaphase chromosomes, with either one or both chromatids of each homolog hybridized to the probe. Similar or higher hybridization efficiency results were obtained for the remaining MAGEL2probes and each of the HIRA and CDC2L1 probes. The metaphase chromosome hybrid ization patterns were identical, indicating that the probes did not contain repetitive sequences. Interphase cell hybridization patterns were also consistent with probes devoid of repetitive or low copy duplicated sequences.

scFISH on human metaphase chromosomes stained with 4‘,6-diamidino-2-phenylindole (DAPI). Probes are labeled with digoxigenin-dUTP and detected with rhodamine-conjugated antibody to digoxigenin. Hybridized chromosomes are indicated by arrows. PanelsA and B show chromosome 15q11.2–specific hybridization of the 3544-bp MAGEL2 probe with (A) and without (B) preannealing with Cot1 DNA. No differences in the respective hybridization patterns are observed. Panels C and D indicate hybridization of chromosome 1p36–specific CDC2L1 probes in the absence of Cot1 DNA. The 4823-bp probe is shown in panel C, and the combined 4823-bp and 4724-bp probes are shown in panel D. Chromosome 1 homologs in both panels show a single region of hybridization. Panels E and F show hybridization of chromosome 22q11.2 HIRA probes (combined 2848-bp, 3344-bp, 3691-bp, and 5170-bp fragments) on cells from a normal individual (E) and an individual with DiGeorge syndrome (F). Panel F shows a hemizygous chromosome 22q11.2 deletion (*).
Multiple probes from neighboring genomic DNA intervals were combined and hybridized to metaphase chromosomes in the absence of Cot1 DNA. Their hybridization patterns were compared to those produced by individual probes from the same region. The individual and combined probes produced similar hybridization patterns, regardless of whether they had been preannealed with purified repetitive DNA. For example, the 4823-bp fragment alone (Fig. 4C) and the combined 4724- and 4823-bp fragments (Fig. 4D) both showed similar hybridization patterns on chromosome 1. Hybridization to other chromosomal locations was not evident, indicating that both probes were free of repetitive sequences. Generally, signals from mixtures of chromosome specific probes were more intense than individual probes, presumably because the chromosomal target was longer. Interestingly, however, target chromosomal length was not the only factor influencing signal intensity. A smaller 2378-bp fragment contained within the 4823-bpCDC2L1 probe and mapping near the chromosome 1p telomere consistently showed stronger hybridization intensity than the similarly sized 2290-bp probe from the MAGEL2 locus in the chromosome 15 centromeric region (data not shown). These hybridization data and sequence analyses indicate that base composition, probe length, and chromosomal location contribute to hybridization signal intensity.
To show that scFISH probes detected known genetic abnormalities, we hybridized the chromosome 22 probes to cells of individuals with DiGeorge syndrome. These individuals were previously shown to have a molecular cytogenetic deletion of chromosome 22q11.2 with a commercially available probe [TUPLE1] for DiGeorge syndrome (Vysis, Inc). Representative hybridizations using a mixture ofHIRA probes spanning 15 kb of chromosomal target DNA in a control individual (Fig. 4E) and an individual with DiGeorge Syndrome (Fig. 4F) are shown. Both copies of chromosome 22 hybridized in cells of the control individual, whereas the probe mixture hybridized to a single chromosome 22 in cells from the individual with DiGeorge Syndrome.
Feasibility of Genomewide Application
To assess the potential for developing scFISH probes for other genomic regions, we analyzed the organization of single-copy sequences on chromosomes 21 and 22 (Table 2). Chromosome 21 contains fewer single-copy intervals than chromosome 22, and the intervals are, on average, shorter. Adjacent intervals tend to be clustered on chromosome 22, with 39% separated by 500–1000 bp. The distributions of interval lengths for both chromosomes are narrow and have wide tails (i.e., leptokurtic). Single-copy intervals ≥2.3 kb (the length of the shortest scFISH probe visualized in this study) are separated, on average, by 29.2 kb on chromosome 21 and by 22.3 kb on chromosome 22.
Distributions of Single Copy Sequence Intervals
Single-copy intervals are not uniformly distributed on chromosome 22, based on their frequencies in 1-mb genomic bins. The centromeric and telomeric regions are more densely populated than the central region; however, the disparity between the different regions is not very marked. The 33-mb bin close to the telomere has the greatest density (one per 8.8 kb), and the 4-mb bin near the centromere has the next highest density (one per 11.1 kb). The 16-–19-mb bins in the middle of the chromosome have the fewest single-copy intervals, averaging one single-copy sequence per 33.3 kb.
We examined the distribution of distances between adjacent ≥2.3-kb single-copy sequences on chromosome 22q. Single-copy segments separated by 1.25 kb–100 kb have a near normal distribution, but densely clustered single-copy intervals (≤1.25 kb) and sparsely populated, single-copy chromosomal regions (≥100 kb) are more prevalent than expected (P<0.0001). The densely clustered single-copy intervals are distributed throughout the chromosome, whereas 22 of 29 sparsely populated genomic regions are scattered throughout a 10.6-mb interval in the middle of the chromosome (bins 8–19-mb). Similarly, chromosome 21q has 46 genomic intervals ≥100 kb that do not contain single-copy segments ≥2.3 kb in length.
To estimate the size of genomic intervals required to develop scFISH probes, we determined the probability of detecting at least one single-copy sequence in overlapping, uniform-length genomic intervals on chromosomes 21q (Fig. 5A) and 22q (Fig.5B). Single-copy segments ≥2.0 kb in length are found in the majority of 100-kb genomic intervals of these chromosomes (96% of chromosome 22q and 88% of chromosome 21q). Increasing the size of the genomic sequence to 150 kb results in 99% coverage of chromosome 22q and 96% coverage of chromosome 21q. Single-copy segments of ≥3 kb are considerably less frequent and are found in 94% of chromosome 22q and 82% of chromosome 21q genomic intervals 218 kb in length. A large proportion of 218-kb genomic intervals did not have single-copy segments ≥4 kb in length on either chromosome 21 (62%) or 22 (24%). Therefore, scFISH probes should be ∼2 kb to ensure comprehensive coverage (at least once per 100–150 kb) of chromosomes 21q and 22q. Assuming that single-copy sequences are similarly distributed on other chromosomes, it should be feasible to develop scFISH probes for molecular cytogenetic analysis of most clinically relevant chromosomal rearrangements.
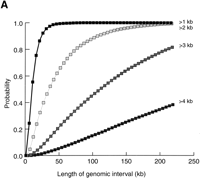
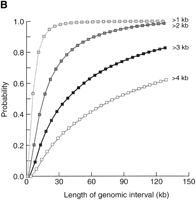
Probability of detecting single-copy segments within genomic domains of varying lengths. Probabilities for single-copy segments of ≥1, ≥2, ≥3, and ≥4 kb are plotted as indicated; (A) chromosome 21q, (B) chromosome 22q. Overlapping genomic intervals were analyzed in increments of 5 kb up to 218 kb; however, only interval lengths up to 130 kb are indicated for chromasome 22.
DISCUSSION
We have masked repetitive elements in genomic sequence contigs to identify single-copy intervals and design probes for chromosomal fluorescence in situ hybridization. Our results indicate that this approach is practical for developing probes from gene-rich regions and that scFISH is likely to be applicable for many regions of the genome. Multiple probes were developed from three chromosomal regions—15q11.2, 22q11.2, and 1p36—all of which in the hemizygous state lead to clinically recognizable syndromes. Deletions of chromosome 15q11.2 lead to two clinically distinct syndromes, Prader-Willi syndrome or Angelman syndrome, depending on the parental origin of the chromosome (Knoll et al. 1989; Nicholls et al. 1989). Deletions of chromosome 22q11.2 lead to DiGeorge syndrome (Fibison et al. 1990; Carey et al. 1992; Driscoll et al. 1992; Consevage et al. 1996); deletions of 1p36, to an aneusomic disorder of mental and physical impairment (Slavotinek et al. 1999). The MAGEL2, HIRA, and CDC2L1 genes are in the critical regions of Prader-Willi/Angelman, DiGeorge, and Monosomy 1p36 syndromes, respectively. We showed localization of all probes to their respective chromosomal regions and confirmed the localization of the HIRA probes to within the DiGeorge syndrome critical region by showing a molecular deletion in cells from such individuals.
scFISH and cloned genomic probes differ in a number of respects. Aside from the absence of repetitive DNA, scFISH probes are enriched for expressed sequences because the genomic intervals from which they were derived are presumed to contain genes implicated in specific disorders. These probes usually contain coding and noncoding sequences, but occasionally consist entirely of single-copy, unexpressed sequence intervals (i.e., the 4100-bp and 2290-bp probes from theMAGEL2 locus). Additionally, scFISH probes can be quickly designed and produced by long PCR. Propagation of conventional FISH probes prepared by recombinant DNA techniques is slower, especially when library screening is taken into account.
The sequence content of scFISH probes is precisely defined, in contrast with unsequenced cloned probes that are commonly used for FISH. Cloned probes may, in some instances, detect low-copy complex repetitive sequence families (for review, see Mazzarella et al. 1998; Ji et al. 2000), which participate in a variety of recurrent chromosomal rearrangements, producing additional signals from hybridization to other related loci. To ensure that FISH probes detect unique sequences, scFISH probe sequences are verified by BLAST analysis. To visualize an individual member of a low copy complex repeat family, however, scFISH probes can be prepared with PCR primers designed to uniquely amplify a specific element (i.e., containing weakly conserved regions of the repeat) and washed at high stringency after hybridization.
Based on our analyses of chromosomes 21q and 22q and on the comprehensive state of the genome sequence, it should be feasible to develop scFISH probes for molecular analysis of most euchromatic chromosomal rearrangements. Adequate resolution can be obtained with probes ∼2 kb in length to detect chromosome breakpoints and gene(s) disrupted by such rearrangements, as has been previously performed by Southern hybridization and occasionally by fiber-FISH, neither of which preserves higher-order chromosome structure (Florijn et al. 1995). By expanding the repertoire of probes available for molecular genetic analysis of chromosomal alterations, probes can be developed to delineate multigene family members, identify and size marker chromosomes, and detect uncommon chromosome abnormalities that could not otherwise be studied (because commercial or research probes are not available). scFISH will also find application in chromosomal analysis of other organisms for which comprehensive catalogs of repetitive sequences have been compiled and genomic sequences of sufficient length are available.
METHODS
Identification of Single-Copy Sequences
We deduced the locations of single-copy probe sequences directly from long contiguous genomic DNA sequences. The locations were determined by software that aligns the sequences of repetitive sequence family members with the target genomic sequence. Comparison of the target sequence with previously determined sequences of repetitive family members can identify and delineate the bounds of repetitive elements within the target. The computer program CENSOR(Jurka et al. 1996) was used to determine the locations of repetitive sequence families in contiguous genomic sequences ≥100 kb in length.CENSOR compares a genomic sequence with a compilation of repetitive sequence families present in multiple copies in the human genome (http://www.girinst.org). This repeat sequence database contains representative and consensus sequences for the majority of repetitive sequence families.
A Perl script (findi.pl) parsed the coordinates of the boundaries of the repetitive segments from CENSOR output and then deduced and sorted the adjacent single-copy intervals by size greater than a parametrized threshold (≥2 kb in most instances). The boundaries of adjacent single-copy intervals were deduced by subtracting one nucleotide position from the upstream boundary of a repetitive element and adding one nucleotide position to the downstream boundary of the previous element. Single-copy intervals with identical upstream and downstream coordinates (1 bp in length) were considered to be adjacent repetitive sequences. Probe sequences were then compared with the human genome sequence database (Altschul et al. 1990) to determine if there was similarity to sequences elsewhere in the genome.
Oligonucleotide primers were selected for PCR amplification of the longest single-copy intervals. A Unix wrapper optimized primer selection by iterating the following parameters for input to the program Prime (Genetics Computer Group): Tm(from 70 → 60o C), G/C composition (from 55% to 40%), and minimum interval length (from 90% to 80% of the length of the single-copy interval).
Probe Generation and Chromosomal In Situ Hybridization
DNA fragments, ranging from 2290 to 5170 bp in length, were amplified by long PCR (Cheng et al. 1994) with LA-Taq as recommended by the manufacturer (Panvera). The amplicons were purified by low-melt temperature agarose gel electrophoresis, followed by chromatography with Micro-spin columns (Millipore), which removed contaminating extension products containing repetitive sequences.
Probe fragments were labeled by nick translation using modified nucleotides such as digoxigenin-dUTP or biotin-dUTP (Roche Molecular Biochemicals). Labeled probes were denatured and hybridized to fixed chromosomal preparations on microscope slides using our previously described conditions (Knoll and Lichter 1994), with the exception that preannealing of the probe(s) with repetitive DNA (such as Cot1 DNA) was not used in a parallel set of hybridizations. Probes from a single chromosome region of ∼100kb were hybridized individually or in combination to remove nonspecific binding. Posthybridization washes were performed at 42°C in 50% formamide in 2xSSC, followed by an additional wash at 39°C 2xSSC and one in 1xSSC at room temperature. Wash stringency was increased, if necessary, to remove hybridization of probes to related sequences elsewhere in the genome. Hybridized probes were detected with a fluorochrome (such as rhodamine or fluorescein) tagged antibody to the modified nucleotide. Chromosome identification was performed by counterstaining the cellular DNA with 4‘, 6-diamidino-2-phenylindole (DAPI). Hybridized chromosomes were viewed with an epifluorescence microscope (Olympus) equipped with a motorized multiexcitation fluorochrome filter wheel. Hybridization patterns on at least 20 metaphases (and 50 to 100 nuclei) were scored for each probe or combination of probes, with and without preannealing to Cot1 DNA. Cells were imaged using a CCD camera (Cohu) and CytoVision ChromoFluor software (Applied Imaging).
Feasibility of Approach for Detecting Single-Copy Intervals
The distribution of single-copy sequences, and thus potential scFISH probes, along a typical chromosome was estimated by analyzing the 21q and 22q sequences. The coordinates of each of the repetitive sequence elements in available chromosome 21 (Hattori et al. 2000), and chromosome 22 sequences (Dunham et al. 1999) were located with theCENSOR program. The intervening intervals were presumed to consist predominantly of single or low copy sequences. The locations and lengths of each intervening interval and the distances separating adjacent intervals were computed with the findi.pl script.
The lengths of genomic sequences required to find scFISH probes exceeding a parametrized length were determined with the Perl scriptprobsc.pl. This program computes the probability of detecting at least one single-copy interval greater than a specified length in every genomic interval on both chromosomes. For each single-copy window length, a range of genomic windows was tested up to 220 kb.
Chromosomal single-copy interval distributions were analyzed withSPSS v. 9.0 (SPSS) to estimate the resolving power of scFISH for genome-wide studies. To indicate deviations from a normal distribution, lengths and distances between intervals were plotted on a log scale and significance was evaluated with the Kolmogarov-Smirnov statistic.
Acknowledgments
The sequence data containing the CDC2L1 locus were produced by the Human Chromosome 1 working group at the Sanger Centre and can be obtained from ftp://ftp.sanger.ac.uk/pub/chr1. The sequence data containing the HIRA locus were produced by the Advanced Center for Genome Technology at the University of Oklahoma and can be obtained from ftp://ftp.sanger.ac.uk/pub/human/chr22. The sequence of the MAGEL2 genomic locus was produced by the Genome Science & Technology Center at the University of Texas Southwestern Medical Center (http://gestec.swmed.edu/). We gratefully acknowledge support from the Katherine B. Richardson and Patton Trusts.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL progan{at}cmh.edu; FAX (816) 753-1307.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.171701.
-
- Received November 27, 2000.
- Accepted March 2, 2001.
- Cold Spring Harbor Laboratory Press








