
The 1.4-Mb CMT1A Duplication/HNPP Deletion Genomic Region Reveals Unique Genome Architectural Features and Provides Insights into the Recent Evolution of New Genes
Abstract
Duplication and deletion of the 1.4-Mb region in 17p12 that is delimited by two 24-kb low copy number repeats (CMT1A–REPs) represent frequent genomic rearrangements resulting in two common inherited peripheral neuropathies, Charcot-Marie-Tooth disease type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsy (HNPP). CMT1A and HNPP exemplify a paradigm for genomic disorders wherein unique genome architectural features result in susceptibility to DNA rearrangements that cause disease. A gene within the 1.4-Mb region,PMP22, is responsible for these disorders through a gene-dosage effect in the heterozygous duplication or deletion. However, the genomic structure of the 1.4-Mb region, including other genes contained within the rearranged genomic segment, remains essentially uncharacterized. To delineate genomic structural features, investigate higher-order genomic architecture, and identify genes in this region, we constructed PAC and BAC contigs and determined the complete nucleotide sequence. This CMT1A/HNPP genomic segment contains 1,421,129 bp of DNA. A low copy number repeat (LCR) was identified, with one copy inside and two copies outside of the 1.4-Mb region. Comparison between physical and genetic maps revealed a striking difference in recombination rates between the sexes with a lower recombination frequency in males (0.67 cM/Mb) versus females (5.5 cM/Mb). Hypothetically, this low recombination frequency in males may enable a chromosomal misalignment at proximal and distal CMT1A–REPs and promote unequal crossing over, which occurs 10 times more frequently in male meiosis. In addition to three previously described genes, five new genes (TEKT3, HS3ST3B1, NPD008/CGI-148, CDRT1, andCDRT15) and 13 predicted genes were identified. Most of these predicted genes are expressed only in embryonic stages. Analyses of the genomic region adjacent to proximal CMT1A–REP indicated an evolutionary mechanism for the formation of proximal CMT1A–REP and the creation of novel genes by DNA rearrangement during primate speciation.
Submicroscopic duplications/deletions represent genomic rearrangements that can be responsible for inherited diseases. These are not visible by conventional karyotype assays and are thus likely to involve rearranged fragments smaller than 1–2 Mb. Disorders with these types of rearrangements may be caused by dosage effects of a single or multiple genes. Inherited diseases resulting from such genomic rearrangement may be categorized as genomic disorders in contrast to classic Mendelian diseases caused by point mutations in the causative genes (for review, see Lupski 1998b; Shaffer and Lupski 2000).
Charcot-Marie-Tooth disease type 1A (CMT1A) is one of the first and best-characterized examples of a submicroscopic genomic disorder. CMT1A is the most common inherited peripheral neuropathy and accounts for 70% of CMT type 1 inherited demyelinating neuropathy (for review, seeLupski and Garcia 2001). Molecular genetic approaches have identified a submicroscopic duplication of the 1.4-Mb genomic region in chromosome band 17p12 in the majority of the CMT1A cases (Lupski et al. 1991;Raeymaekers et al. 1991; Wise et al. 1993; Nelis et al. 1996; Roa et al. 1996). A submicroscopic deletion of the same region results in hereditary neuropathy with liability to pressure palsy (HNPP), a distinct form of inherited peripheral neuropathy with episodic and milder manifestations (Chance et al. 1993, 1994). The CMT1A duplication and HNPP deletion represent products of unequal crossing over and a reciprocal recombination between flanking 24-kb homologous sequences termed CMT1A–REPs (Lupski 1998a). Subsequently, a gene encoding PMP22, a major component of the peripheral nervous system myelin, was mapped in the middle of this 1.4-Mb region (Matsunami et al. 1992; Patel et al. 1992; Timmerman et al. 1992; Valentijn et al. 1992). Several lines of evidence indicate that gain of one copy of PMP22 is responsible for CMT1A, whereas loss of one copy of PMP22results in HNPP through a PMP22 gene dosage effect as the mechanism for these disorders (Lupski et al. 1992).
Although duplication and deletion of PMP22 is the event responsible for CMT1A and HNPP, respectively, as many as 30 to 50 other genes may be contained in this 1.4-Mb region on the basis of its genomic size (Murakami et al. 1997b). A question remains as to why onlyPMP22 is dosage sensitive, whereas other genes in the region are apparently not. In addition, the clinical phenotypes of patients having the same 1.4-Mb duplication are quite variable. A formal possibility exists that minor dosage effect of genes other thanPMP22 in this 1.4-Mb region somehow contribute to the variability of phenotypic manifestations or a combination of phenotypes (e.g., CMT + connective tissue disorder). Furthermore, there are rare case reports of smaller duplications (Ionasescu et al. 1993; Palau et al. 1993; Valentijn et al. 1993) or deletion (Chapon et al. 1996), raising the question as to whether such rare recombination events are mediated by other repeat units in this region.
To characterize the genomic architecture of this region, we constructed PAC and BAC contigs and produced a finished sequence across this 1.4-Mb interval. We defined a 1,421,129-bp genomic interval as the CMT1A duplication/HNPP deletion region. Here we report the identification of low-copy number repeats (LCRs), the comparison of genetic and physical maps, the identification and characterization of genes, and a mechanism for the evolution of new mammalian genes by DNA rearrangements.
RESULTS
Sequencing the 1.4-Mb CMT1A Duplication/HNPP Deletion Region
A contig of overlapping bacterial clones was constructed on the basis of marker content by use of pre-existing and newly generated STSs. Restriction fragment fingerprinting (Marra et al. 1997) verified the order of clones within the contig and identified a set of minimally overlapping BAC and PAC-tiling path of clones for genomic characterization. Individual clones were subjected to shotgun sequencing, assembly, and finishing. A path of 12 overlapping clones contains the complete region bounded by the CMT1A–REPs, and this is part of a larger 15-clone path analyzed in this study (Fig.1). Previously, we have predicted the size of this genomic region to be 1.5 Mb on the basis of physical mapping data obtained by pulsed-field gel electrophoresis (PFGE) and Southern blotting analyses (Pentao et al. 1992). Our completed sequence indicates that the entire region from the first nucleotide of the proximal CMT1A–REP to the last nucleotide of the distal CMT1A–REP is 1,421,129 bp.

The genomic sequence map of the CMT1A duplication/HNPP deletion region in 17p12. The top solid horizontal line represents the genomic sequence of the CMT1A/HNPP region in the centromere to telomere orientation. Position 0 is assigned to the first base of the proximal CMT1A–REP and vertical markings are placed every 100 kb for reference. The STR polymorphic genetic markers are shown above. Shaded horizontal boxes below depict the large insert clones used to derive genomic sequences. Clones are identified by their individual names and GenBank accession numbers. Proximal and distal CMT1A–REPs are shown as vertical blue boxes, and newly identified low copy repeats (LCRA1, LCRA2, LCRB) are represented as vertical red bars. Arrowheads indicate the orientation/direction of each repeat unit. Underneath are shown known genes (green), predicted genes (purple), and pseudogenes (black), with arrows pointing in the direction of transcription. (MITE and HSMAR2-PMP22) mariner transposon-like elements; (CYPAP) cyclophilin A pseudogene; (60SRPL9P) 60S ribosomal protein L9 pseudogene; (60SRPL23AP) 60S ribosomal protein L23A pseudogene; (40SRPS18P) 40S ribosomal protein S18 pseudogene.
Repetitive Elements
RepeatMasker indicates that high copy number retrotransposable elements and simple tandem repeats (STRs) account for 43.37% of the entire CMT1A/HNPP region (Table1). The repetitive elements consist of 9.97% Alu sequences and 13.43% LINE1 elements, which is comparable in distribution with that of chromosome 21, but in contrast to that of chromosome 22, which contains 16.8% of Alusequences and 9.73% of LINE1 elements (Dunham et al. 1999; Hattori et al. 2000).
The Interspersed Repeat Content of the CMT1A/HNPP Region
There is a mariner insect transposon-like element 140-kb centromeric to PMP22, termed HSMAR2–PMP22 (Fig 1). This mariner element is interrupted by an insertion of anAlu element, indicating that it is no longer active. However, we observed both 5′ and 3′ inverted terminal repeats (ITRs), suggesting that this mariner element has the potential to act as acis-acting substrate to promote double-strand DNA breakage (Reiter et al. 1996, 1999).
We identified 53 STRs with repeating units >11. Nine STRs (D17S793, D17S261, D17S122, D17S1357, D17S1356, D17S839, D17S1358, D17S955, and D17S921) were mapped previously to this region, two (D17S918 and D17S900) were mapped to the region but not known to be within the CMT1A/HNPP interval, and forty-two represent newly identified potential polymorphic markers. The new STRs include 26 dinucleotide [21 (CA)n, 2 (GA)n, 1 (TA)n, 1 (TA)n(CA)n, and 1 (TG)n(GA)n], 2 trinucleotide [2 (CAA)n], 10 tetranucleotide [6 (TTTA)n and 4 (TTTC)n], and 4 pentanucleotide [1 (TTTTC)n, 1 (CAATA)n, 1 (CGATA)n, and 1 (TTTTA)n] elements. Fifteen of these STRs have been shown to reveal significant polymorphic variation in different ethnic populations (Badano et al. 2001).
Low Copy Repeats: An 11-kb Element
In addition to the previously defined CMT1A–REPs (24,011 bp of 98.7% nucleotide identity, Reiter et al. 1997), other low copy repeats were identified (Fig.1). LCRA1 and LCRA2, located 32-kb centromeric and 140-kb telomeric to the distal CMT1A–REP in inverted orientaion, are highly similar 11-kb low copy number repeat segments. We also found a 4-kb truncated copy of this repeat, termed LCRB, ∼180 kb centromeric to the proximal CMT1A–REP (Fig. 1). Therefore, one copy of this repeat is located within the 1.4-Mb region and the other two are located outside of this region. LCRA1 and LCRA2 are highly similar throughout the 11 kb (97% identity), whereas LCRB aligns only with a 4-kb interior portion (95% identity to LCRA1) (Fig.2A). Further sequence comparisons revealed one small region (132 bp) that represents DNA rearrangements between these LCRs (Fig. 2A). LCRA1 contains three contiguous fragments (25, 89, and 18 bp) that involve small tandem repeat units (14- and 9-bp monomer). The corresponding region in LCRA2 contains a duplication of the 25-bp monomer as well as a deletion of the 18-bp fragment, probably resulting from polymerase slippage at the 14- and 9-bp repeat units flanking these 25- and 18-bp fragments, respectively, in LCRA1. Furthermore, the recombination breakpoint of the LCRB is located in this small region between the 14- and 9-bp repeat units, resulting in truncation of the 89-bp fragment and loss of the 25-bp fragment. No 18-bp deletion was found in the LCRB. This genomic evidence indicated that the LCRA1 is likely the progenitor and the other two LCRs are derivatives of LCRA1. A duplication event that results in LCRB may have been followed by another duplication that generated LCRA2.
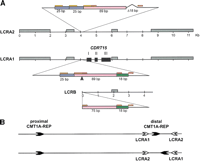
Genomic structure of LCRA1, LCRA2, and LCRB. (A) The genomic architecture of three LCRs is shown. High copy number retrotransposable elements including Alu, L1, MaLR, and MER2 type DNA elements, which are conserved between these LCR, are boxed in gray. Each exon ofCDRT15 is shown as a solid black box. A small genomic region in the 5′ upstream of CDRT15 exon I representing DNA rearrangements (enlarged) between the three LCRs. The LCRA1 has a 132-bp region that is further divided into 25 bp (blue), 89 bp (pink) and 18 bp (green) segments. There are short tandem repeat sequences, 14 bp (orange) and 9 bp (red), flanking these subdivided fragments. In comparison with the same region in the LCRA2, the 25-bp monomer is tandemly duplicated and the 18-bp fragment is deleted in LCRA2. Furthermore, the distal boundary of 4.4-kb LCRB is located in this short genomic region between 14- and 9-bp repeat (arrow). These 18 bp are present in LCRB. (B) Hypothetical inversion involving LCRA1 and LCRA2 results in flipping of distal CMT1A–REP. The proximal and distal CMT1A–REPs are depicted by thick black arrows with their relative orientation given by the directions of arrows. LCRA1 and LCRA2 are depicted as open arrows. The genome architecture as determined by sequencing this specific genomic library alone placed the CMT1A–REPs in a direct orientation and thus the region in between is susceptible to duplication/deletion. Below is shown the hypothetical orientation of CMT1A–REPs if an inversion occurs via homologous recombination using LCRA1 and LCRA2 as substrates. Note that this orientation of CMT1A–REPs will prevent the formation of duplication/deletion event (Lupski 1998b).
Searches of the high throughput human genome sequence revealed the presence of multiple copies of this LCR in the genome. After elimination of the highly repetitive 4.4-kb flanking sequences from this 11-kb fragment, BLAST searches with the 6.6-kb region identified 29 BAC clones assigned to 9 different chromosomes; 1, 4, 8, 9, 11, 13, 16, 17, and 22 (data not shown). Electronic PCR analyses (Schuler 1998) of each BAC clone showed STSs from multiple chromosomes matching a single BAC sequence, whereas the 11-kb LCRA1 only contains a chromosome 17-specific STS, suggesting the repeat structures involving these loci in the genome are complex. Further mapping and characterization are required to elucidate the nature of these repeat structures involving multiple loci in the genome.
BLAST searches of this 6.6-kb region against the human EST database revealed a number of clones homologous to this portion of the LCRA1 low copy repeat. There are two different genes or groups of genes; one homologous to the 3kb–4kb region from the centromeric side (named CDRT15, see details in the following section) and the other to the 4.5–6.3-kb region. Further database searching revealed that the latter is a processed pseudogene of KIAA1511, which encodes a protein of unknown function and maps to chromosome 1 (GenBank accession no. AB040944). Interestingly, ESTs belonging to the former group have various levels of homology, suggesting that these ESTs may be transcribed from multiple loci in the genome. Further sequence comparison of these EST clones to the genomic sequence database mapped them to at least nine different genomic loci.
Comparison between the Physical and Genetic Maps
In previous efforts to identify the CMT1A gene by linkage analysis, the CMT1A region was estimated to be much larger than 1.4 Mb on the basis of the genetic distance between linked markers (Patel et al. 1990; Timmerman et al. 1990). However, subsequent physical mapping with PFGE and YAC-based STS content mapping revealed a physical size of 1.5 Mb (Pentao et al. 1992). One hypothesis to explain the observed discrepancy between genetic and physical distances has been that a potential recombination hotspot exists within the CMT1A genomic region in addition to the positional recombination hotspot located within CMT1A–REP (Reiter et al. 1996). To evaluate the actual recombination frequency, we systematically compared the genetic map and genome sequence-based physical map of the CMT1A duplication/HNPP deletion region by integrating the Marshfield genetic mapping data into our physical map. Eight polymorphic microsatellite markers (D17S900, D17S921, D17S955, D17S839, D17S918, D17S122, D17S261, and D17S793) were found in both the Marshfield genetic map and genomic sequence from the 1.4-Mb region (Broman et al. 1998). Of these, two markers (D17S900 and D17S918) were not mapped inside this region in the previous physical maps (Murakami and Lupski 1996; Boerkoel et al. 1999). Three markers identified previously in the CMT1A region (D17S1356, D17S1357, andD17S1358) were not included in the Marshfield study (Blair et al. 1995).
We generated a genetic/physical map correlation (Fig.3A) and compared it with the flanking 1.5-Mb regions. Physical distances in the proximal regions include estimates based on BAC physical mapping data at 100-Kb resolution on the centromeric side (J.R. Lupski and B. Birren, unpubl.) and fully finished sequence on the telomeric side. These genetic/physical map comparisons indicate that the recombination frequency of an at least 4.5-Mb region including the CMT1A duplication/HNPP deletion region is low in males. In sharp contrast, this region recombines frequently in females. The cM/Mb ratio of the entire 4.5-Mb region is 5.5 for female, 0.67 for male, and 3.3 for the sex-averaged map. As a result of this contrast, this region has a high female/male recombination frequency ratio, which is steeply increasing toward the centromere (Fig. 3B). Neither CMT1A–REP regions nor the entire CMT1A/HNPP region have a higher recombination frequency than flanking regions. The 820-kb region between D17S1843 and D17S918, which spans the proximal CMT1A–REP, revealed no recombination in the families examined in both male and female meiosis (Broman et al. 1998). There is also a low recombination region in both sexes telomeric to distal CMT1A–REP for >1 Mb.
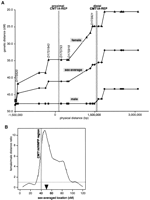
Sex-specific recombination frequencies in the CMT1A/HNPP genomic region. (A) The relationship between genetic and physical distance. The STR markers in the 17p12 CMT1A/HNPP region from the Marshfield genetic map were aligned to the nucleotide sequence-based physical map. The marker order is as follows: centromere-D17S1794-D17S620-D17S2196-D17S1857-D17S953-D17S1843 -D17S793-D17S122-D17S918-D17S839-D17S955-D17S921-D17S900 -D17S922-D17S1856-D17S947-D17S936-D17S639-D17S799-D17S1808-D17S1803-telomere (markers within the CMT1A/HNPP genomic region are underlined). Both CMT1A–REPs are shown as hatched bars. As the sequencing of the centromeric side of the map has not yet been completed, the physical distance was calculated on the basis of the BAC contig map (K. Inoue, K. Dewar, N. Katsanis, L.T. Reiter, E.S. Lander, K.L. Devon, D.W. Wyman, J.R. Lupski, and B. Birren, unpubl.). (B) Female/male distance ratio (vertical axis) is plotted along the sex-averaged genetic map (horizontal axis). The histogram was obtained from the Marshfield Center for Genetics (http://research.marshfieldclinic.org/genetics/Map_Markers/maps/IndexMapFrames.html). The CMT1A/HNPP genomic region is shown as a shaded vertical bar. (▾) The predicted position of the centromere.
Genes in the 1.4-Mb Region
Sequence analysis was performed by the use of NIX (nucleotide identification of unknown sequences), which incorporates a number of independent gene prediction tools (Fig. 1; Table2). Each gene was further characterized by additional database searches and expression analyses. We categorized the genes into three groups; (I) genes for which we have biological evidence including cDNA sequences, gene structures, similarity to other genes, or multiple spliced ESTs, matching gene predictions with complete gene structure; (II) predicted genes with limited information such as multiple EST matches and/or predicted exonic structures, but complete gene structural information is not available, and; (III) pseudogenes. Overall, we identified 21 genes or predicted genes (Groups I and II) in this region.
Summary of the Genes, Predicted Genes, and Pseudogenes in the CMT1A/HNPP Region
Genes
Of the eight genes in this group, four are known: HREP, PMP22, HS3ST3B1, and COX10. Of these,HS3ST3B1 is the only gene newly mapped to this region.COX10 and HREP are located in the CMT1A–REP regions in which complete sequence data were available previously (Reiter et al. 1997; Kennerson et al. 1997, 1998; Murakami et al. 1997a). We thus describe the genomic structures of PMP22 and HS3ST3B1in further detail. Four previously unknown genes were also identified,NPD008/CGI-148, tektin3 (TEKT3),CDRT1 (CMT1A duplicatedregion transcript 1), and CDRT15.
PMP22
PMP22, the gene responsible for CMT1A and HNPP, has four coding exons and two alternatively utilized exons I (Suter et al. 1994;Sabéran-Djoneidi et al. 2000). PMP22 spans 35 kb and is transcribed toward the telomere (Fig. 4A). One trinucleotide repeat sequence was found in intron 3, which matched a previously known STR, D17S918. This STR contains 12 CAG repeats.
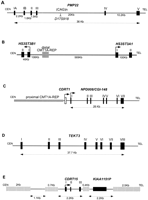
Genomic structure of the genes in the CMT1A/HNPP region. (A)PMP22. Arrowhead indicates marker D17S918, which contains polymorphic CAG repeats. Distances for each intron are shown at bottom, whereas individual exons are numbered attop. We hypothesized that in rare families with CMT accompanied by anticipation, it may be related to an expanded allele of this triplet repeat. To test this hypothesis, we obtained DNA samples from one such family for which the disease locus mapped to 17p11.2–17p12 by linkage analysis (Kovach et al. 1999). We examined the number of CAG repeat in members of this family, but failed to identify any expansion of the triplet repeats in affected individuals (data not shown). A point mutation in PMP22 was subsequently found to segregate with the disease phenotype in this family (Kovach et al. 1999). (B) HS3ST3B1 and HS3ST3A1. HS3STB1 is located inside the CMT1A/HNPP genomic region, whereas HS3STA1 is 569-kb telomeric to the distal CMT1A–REP. Each exon of the two genes is indicated as a box, and arrows show the direction of the transcription. (C) CDRT1 andNPD008/CGI-148. The horizontal shaded rectangle indicates the proximal CMT1A–REP. An open box shows the single exon ofCDRT1 and solid boxes indicate seven exons ofNPD008/CGI-148 that span 26 kb. (D)TEKT3 contains eight exons (solid boxes) including an untranslated first exon. The putative initiating methionine is in exon II. The exon/intron boundaries from exons III to VI, which were not determined by database analyses, were experimentally confirmed by RT–PCR and sequence analyses using single-stranded testis cDNA as the template DNA. (E) CDRT15 in the 11-kb low-copy repeat unit, LCRA1. Hatched horizontal bars indicate repetitive elements. Pseudogene for KIAA1151 is also shown.
HS3ST3B1
The cDNA sequence for HS3ST3B1 (heparan sulfate D-glucosaminyl) 3-O-sulfotransferase 3B1) was described previously, but the genomic structure was unknown (Shworak et al. 1999). HS3ST3B1 encodes a 390 amino acid enzyme that catalyzes sulfation of heparan sulfate (Liu et al. 1999) and contains two coding exons that are separated by a 43-kb intron (Fig. 4B). HS3ST3B1is part of a gene family that includes HS3ST1, HS3ST2, HS3ST3A1, HS3ST3B1, and HS3ST4 (Shworak et al. 1999). BothHS3ST3A1 and HS3ST3B1, which are highly similar within their sulfotransferase domains (99.2%), map to 17p12. HS3ST3A1 is located ∼700-kb telomeric to HS3ST3B1, and these genes flank the distal CMT1A–REP (Fig. 4B). HS3ST3A1 also contains two coding exons separated by a large 100-kb intron, but is transcribed in the opposite direction. Nucleotide sequence analysis of the 100-kbHS3ST3A1 intron and the 43-kb HS3ST3B1 intron revealed no homology, suggesting that these genomic regions are not conserved between the two genes, and thus, if these genes arose through duplication, the event is evolutionarily very ancient.
NPD008/CGI-148
This transcript has a 615-bp ORF, encoding a predicted 205 amino acid protein. The structure of this gene is shown in Figure 4C. The cDNA sequence reveals an almost complete match with two genes in the database, NPD008 (GenBank accession no. AF223467) andCGI-148 (GenBank accession no. AF151906). NPD008 was isolated from pituitary glands, whereas CGI-148 was reconstructed by a comparative EST database search between human andCaenorhabditis elegans (Lai et al. 2000). Ubiquitous expression was observed by Northern blotting and RT–PCR analyses (Fig.5A,B), but embryonic tissues showed higher expression levels, except for the brain. Database searches identified putative orthologs in various species, including Drosophila melanogaster, C. elegans, Schizosaccharomyces pombe, Saccharomyces cervisiae, and Arabidopsis thaliana, but no information is available with regard to function. A mouse ortholog was reconstructed from EST sequences and was found to encode a predicted protein of 205 amino acids with 91% identity to the human protein. The NPD008/CGI-148 gene product is likely a membrane-bound protein with three possible transmembrame domains. The orthologs in other species are likely to have a similar structure. Through GenBank searches, we also identified three processed pseudogenes on 2p13, 7q21–7q22, and 16p13.
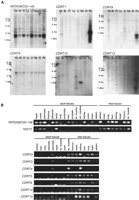
Expression studies of genes identified in the CMT1A/HNPP region. (A) Multiple-tissue Northern blot analyses. Tissues are indicated at top of each lane (He) Heart; (Br) brain; (Pl) placenta; (Lu) lung; (Li) liver; (Mu) skeletal muscle; (Ki) kidney; (Pa) pancreas. Marker sizes are 9.5, 7.5, 4.4, 2.4, and 1.35 kb. (B) Multiple-tissue RT–PCR analyses. NP008/CGI-148 is expressed in a wide variety of tissues. A 2-kb major transcript and two minor transcripts are observed. TEKT3 revealed no expression by Northern blotting (data not show), but high expression in testis by RT–PCR. Faint expression is also observed in both ovary and pancreas by RT–PCR. Fetal tissues reveal low level but ubiquitous expression.CDRT1 shows a major 2-kb and a minor 1-kb transcripts in pancreas. Faint expression is also identified in the heart.CDRT8 reveals two short transcript (0.7 and 1 kb) in the pancreas. A 3-kb transcript of CDRT9 represents low-level ubiquitous expression. CDRT10 is expressed in the skeletal muscle as a 1.3-kb transcript. CDRT12 reveals a 2.8-kb transcript in the pancreas at very low expression levels. CDRT2, CDRT3, CDRT4, CDRT5, CDRT6, CDRT14, and CDRT15 reveal no expression in adult tissues by Northern blotting (data not shown) and RT–PCR analyses, but show obvious expression in fetal tissues.
TEKT3
TEKT3 (Tektin3), located 50-kb centromeric toPMP22, spans 37.7 kb (Fig. 4D). Its eight exons encode a 490 amino acid protein with significant homology to the tektin protein families. The closest homology was to the sea urchin protein, tektin A1, suggesting that this gene is likely to encode a human ortholog for tektin A1, termed TEKT3. As observed in other members of the tektin family, TEKT3 also has a highly conserved tektin domain,RSNVELCRD (underlined residues were conserved in TEKT3) (Norrander et al. 1998; Iguchi et al. 1999). Although Northern blotting analysis failed to show expression in an 8-tissue panel (data not shown), extensive RT–PCR-based expression studies revealed that TEKT3 is primarily expressed in adult testis with low-level widespread expression observed in embryonic tissues (Fig. 5B).
CDRT1
CDRT1 is located 1.3-kb telomeric to proximal CMT1A–REP (Fig. 4C). Multiple human and mouse EST alignments reveal a single exon gene encoding a 243 amino acid protein with unknown function. The upstream 1.3-kb region has weak but potential promoter sequence motifs estimated by the promoter prediction programs TSSW and NNPP. Northern blotting identified a major 2-kb and a minor 1-kb transcript in the pancreas and a faint 2-kb transcript in the heart (Fig. 5A). Further evolutionary analysis of this gene is described in a subsequent section.
CDRT15
CDRT15 is located within the LCRA1. The 778-bp cDNA sequence is divided into three exons, encoding an 188 amino acid protein of unknown function (Fig. 4E). As mentioned above, there are at least eight paralogous copies of this gene in the human genome. Submitted sequences include one full-length cDNA clone encoding an unknown protein (GenBank accession no. AF038169) and numerous partial sequences. We reconstructed complete coding cDNA sequences by aligning these ESTs with each other. At least three cDNA clones were found to contain ORFs with possible exon/intron structures. Interestingly, they have insertion/deletion mutations that result in frameshifts of the ORF, thus encoding totally different proteins; others have insertions/deletions that appear to result in early termination. It is not clear which gene copies are producing functional proteins and which are transcribed pseudogenes.
Predicted Genes
We identified 13 predicted genes (Fig. 1; Table 2). Each of these has incomplete information to determine full-length cDNA sequence. However, substantive evidence, including matching UniGene clusters, matching ESTs with intron structure, and significant scores by gene prediction programs, suggest these represent bona fide genes. Interestingly, Northern blotting analyses of these genes by use of an adult tissue panel revealed minimal expression, whereas RT–PCR analysis indicated substantial expression in embryonic tissues (Fig.5). Results of the database and expression analyses for these 13 genes are summarized in Table 2.
Pseudogenes
Six pseudogenes were identified in the CMT1A/HNPP region (Fig. 1; Table 2). Each locus reveals evidence for absent introns and disrupted coding sequence by mutations, suggesting that they are processed pseudogenes. The pseudogene for cyclophilin A (CYPAP) revealed deletion of a region corresponding to the first 180 bp of cDNA sequence. The pseudogene for KIAA1164 showed deletion for the first 2 kb of original 4 kb cDNA, inversion of a 1-kb region, and insertion of an L1 element.
Evolution of New Genes by DNA Rearrangement During Speciation: Origin of HREP and CDRT1
Database searches to identify mouse orthologs of human genes in this region provided evidence of an additional ancestral rearrangement with functional consequences. Searches with human CDRT1 sequences identified mouse ESTs with coding sequences extending 5′ upstream from the initiation site for the human gene (Fig.6A). Human sequence corresponding to this 5′ extension is not found in the genomic sequence from theCDRT1 region. In fact, the mouse EST sequences that extend 298-bp 5′ from the start of the human CDRT1 gene do not match any sequence in the human genome. However, additional sequences further 5′ in the mouse EST contig show similarity to the human HREPgene. The human HREP gene is located centromeric to the proximal CMT1A–REP and, like the human CDRT1 gene, is transcribed in the telomeric direction, ending within the proximal CMT1A–REP (Kennerson et al. 1997, 1998) (Fig. 1). In searching for a mouse ortholog for HREP, we identified a 759-bp continuous fragment of mouse HREP partial mRNA sequence. The first 269 bp of this sequence aligns with the human cDNA and corresponds to human exons IV and V. However, the remainder of the mouse mRNA does not align with human HREP exon VI, but instead the sequences at the 3′ end of this mouse HREP EST contig contain CDRT1sequences. Exon VI of human HREP is located inside the proximal CMT1A–REP and utilizes complementary sequence ofCOX10 pseudoexon VI. Mice do not have the proximal CMT1A–REP; the proximal CMT1A–REP appeared during primate speciation between gorilla and chimpanzee (Kiyosawa and Chance 1996; Reiter et al. 1997;Boerkoel et al. 1999; Keller et al. 1999). These data suggest that in the mouse, sequences corresponding to human HREP andCDRT1 are part of a single gene. The fact that 298 bp from within the mouse ortholog of HREP does not match genomic sequence on either side of the proximal CMTA1–REP suggests that the primate progenitor to human lost some genome sequence when the proximal CMT1A–REP integrated into this region (Fig. 6B).
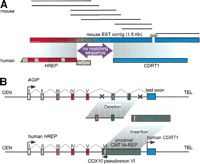
Gene evolution surrounding the proximal CMT1A–REP region. (A) A comparison of EST contigs between mouse and human. Eight mouse ESTs (shown as solid horizontal bars; GenBank accession nos. AI606691,AA089107, AA982328, AI181334, AI882388, AI429210, AI447154, andAI506955, from top to bottom) were reconstructed into a 1.5-kb contig with a 24-bp gap (horizontal rectangle with gradient colors) that aligns with two human genes, HREP (the numbers represent each exon of HREP; the exon VI does not align with the mouse EST contig) and CDRT1. Between the alignment of these two genes, there is a 269-bp region in the mouse clone that does not match any human sequence (purple arrow). The conceptual translation of this region does not identify a known protein functional motif. (B) A model for the evolution of new genes and the genomic structure surrounding the proximal CMT1A–REP region. Topfigure represents the genomic structure of a hypothetical ancient geneAGIP (Ancestral Gene before theIntegration of Proximal CMT1A–REP) modeled in mice. One or more exons originally contained in the AGIPare predicted to be lost by the integration of the proximal CMT1A–REP.Bottom figure shows human genomic structure in whichHREP and CDRT1 are separated by the inserted proximal CMT1A–REP (dark rectangle). The pseudoexon of COX10 is utilized as the last exon of HREP from the opposite direction (green box).
DISCUSSION
Human 17p12 is a genomic region prone to DNA rearrangement (the CMT1A duplication and HNPP deletion) and has undergone relatively recent evolutionary changes during primate speciation (the 24-kb duplicated CMT1A–REPs). Although extensive studies have been performed to elucidate the molecular mechanism for the CMT1A duplication and HNPP deletion, an unequal crossing-over event via homologous recombination utilizing the flanking CMT1A–REPs as substrates, less information has been available for the 1.4-Mb CMT1A/HNPP genomic region between the CMT1A–REPs (Murakami and Lupski 1996; Murakami et al. 1997b; Boerkoel et al. 1999). The finished genomic sequence of this 1.4-Mb region has allowed the elucidation of the genes within the genomic interval and has provided information regarding the genomic architecture of the CMT1A/HNPP region. Our analyses uncovered new LCRs, revealed male-specific reduced recombination, identified novel genes, and shown a mechanism for the evolution of new genes through DNA rearrangement. Our findings suggest that the human genome is in a state of flux with DNA rearrangements apparently responsible for a significant amount of genomic evolution.
LCRs
Large genomic rearrangements mediated by LCR units are associated with a number of human genomic disorders (Lupski 1998b; Shaffer and Lupski 2000). In the CMT1A/HNPP region, in addition to the previously reported CMT1A–REP (Pentao et al. 1992; Reiter et al. 1996,1997), we have identified three copies of a novel LCR, LCRA1, LCRA2, and LCRB. Interestingly, the genomic organization of LCRA1 and LCRA2 consists of inverted repeats flanking the 200-kb region containing the distal CMT1A–REP (Fig. 1). This genomic structure may allow flipping or inversion of the 200-kb genomic fragment in between, thus resulting in the CMT1A–REPs having an inverted orientation (Fig.2B). Such a genomic arrangement may prevent the interchromosomal unequal crossing over that results in CMT1A duplication and HNPP deletion, making such individuals less susceptible to de novo duplication/deletion. This hypothesis is directly testable by determining the CMT1A–REP orientation in the parent of origin for the de novo rearrangement.
A nucleotide sequence comparison between these LCRs revealed that the LCRA1 is likely a progenitor and the other two arose from subsequent duplication events. Two features indicate that the LCRB was probably generated first by local duplication followed by another duplication event to generate LCRA2 from LCRA1. First, the 18-bp deletion only exists in LCRA2 and the sequence homology between LCRA1/LCRB is lower than that between LCRA1/LCRA2. Secondly, a corresponding copy ofCDRT15 in LCRA2 has premature termination and thus is likely a pseudogene of CDRT15.
Multiple copies of LCRs are distributed throughout the human genome. Some BAC clones containing these LCRs map to the Smith-Magenis syndrome (SMS) region on 17p11.2. SMS–REP is a large (>200 kb) low copy region-specific repeat that acts as an homologous recombination substrate and is responsible for a large (∼4 Mb) genomic deletion and duplication associated with human disorders (Chen et al. 1997; Potocki et al. 2000). Six copies of the LCRs were also mapped in 22q11.2, but not in the chromosome 22-specific LCRs (Dunham et al. 1999). Therefore, this LCR family manifests complex divergence throughout the human genome. Because copies of this LCR family are located close to the recombination breakpoints of SMS in 17p12, this LCR family may potentially be involved in the mechanism generating other genomic disorders.
Furthermore, these genome-wide repeat units also involve a gene family that reveals multiple transcripts from different loci. At least three copies of the transcript with no premature termination have been isolated. Further characterization of the sequences of these genomic loci as well as determination of the function of CDRT15 and its paralogs will clarify the complicated structure of these LCRs.
Comparison of Genetic and Physical Maps of the CMT1A Duplication/HNPP Deletion Region
We hypothesized previously that the mariner transposon-like element MITE, which is located ∼500 bp proximal to the preferential region for strand exchange or hotspot for unequal crossing over in the CMT1A–REPs, may promote double-strand DNA breaks and stimulate the homologous recombination (Reiter et al. 1996, 1998). Multiple studies from CMT1A duplication and HNPP deletion patients in different world populations confirm a positional hotspot for recombination within an ∼500-bp region of the 24,011-bp homologous CMT1A–REPs (Kiyosawa et al. 1995; Lopes et al. 1996; Reiter et al. 1996; Timmerman et al. 1997;Yamamoto et al. 1997; Chang et al. 1998). It has been suggested that CMT1A–REPs may also mediate high-frequency homologous recombination of this region at a genomic level.
To investigate this latter hypothesis, we examined the relationship between genetic and physical distances using 21 known STS markers that span this portion of the genome (Fig. 3A). Although we expected increased recombination frequency at some specific cis-acting sequence, such as CMT1A–REPs or HSMAR2–PMP22, there is no significant change in the recombination frequency throughout the region. Instead, we observed evidence for reduced recombination in the 820-kb region between D17S1843 and D17S918 that contains the proximal CMT1A–REP and two of three HSMAR2 elements. These data indicate that the HSMAR2 elements may not increase the frequency of the recombination in the germ line, or the resolution and sensitivity to detect their effect on recombination ratio may be below the lower limit of detection in this study.
Interestingly, in male meiosis, the genomic region with low recombination frequency extended beyond the CMT1A region in both the proximal and distal directions. As shown in chromosome 7, high female/male distance ratio in the genetic versus physical map is likely the result of reduced recombination in males, not of enhanced recombination in females (Broman et al. 1998). There was no recombination identified in the male meiotic map betweenD17S921 and D17S620 (∼3 Mb), whereas in females this same physical distance revealed a 20-cM genetic distance. This reduced male recombination frequency may result in an extended region of two allelic chromosomes without crossing over or synapse formation in meiosis. Such an absence of synapse formation could in turn allow the chromosomes to slip on each other, thus enabling an unequal crossover involving the tandem repeat units, CMT1A–REPs. On the other hand, frequent interchromosomal equal crossovers may provide anchors to prevent chromosomal slipping and reduce the chance of unequal crossovers between the proximal and distal CMT1A–REPs. In support of this hypothesis, de novo CMT1A duplication events occur 10 times more frequently in males than females (Palau et al. 1993; Lopes et al. 1997). Therefore, we hypothesize that one of the mechanisms for the male sex preference in de novo CMT1A duplication may result from the male sex-specific low recombination frequency throughout the region. Interestingly, in the studies of human trisomies, significant reduction of genetic recombination was observed in the trisomy-generating meiosis, and it was suggested that absence of pairing and/or recombination contributes to nondisjunction (Lamb et al. 1996). In the context of the hypothesis that decreased recombination may increase the unequal crossover at the proximal and distal CMT1A–REPs, individuals with reduced meiotic recombination may have an increased propensity to generate unequal reciprocal recombination products.
Han et al. (2000) reported recently that the frequency of unequal crossover between the proximal and distal CMT1A–REPs is almost identical to that of the average equal crossover in the human genome by use of sperm DNA analysis. This hypothesis also indicates that the CMT1A–REPs do not contain a genomic recombination hotspot for the unequal crossover. In the same study, Han et al (2000) localized the recombination breakpoint in the same hotspot identified previously by the analysis of patient DNA. Together with the fact that the CMT1A–REPs do not contain a genomic hotspot for equal crossover according to the comparison of the genetic and physical maps in this study, the hotspot in the CMT1A–REP should be defined as a hotspot for the position preference, not for recombination frequency (Han et al. 2000).
Genes in the CMT1A Duplication/HNPP Deletion Region
In the 1.4-Mb CMT1A duplication/HNPP deletion region, we identified five genes and 13 predicted genes in addition to three previously mapped genes. The current estimated average number of human genes per Mb is between 9.6 and 12.9 (International Human Genome Sequence Consortium 2001). Previous studies suggested that chromosome 17 is gene-rich by a factor of 1.44 (Deloukas et al. 1998), which increases the estimated number of the genes on chromosome 17 to be between 13.8 and 18.6 per Mb. The combination of the eight confirmed and 13 predicted genes within this 1.4-Mb region yields a density of 15 genes/Mb, well within this estimate.
In addition to PMP22, we mapped one previously characterized and two uncharacterized genes to this region, HS3ST3B1, NPD008/CGI-148, and TEKT3. HS3ST3B1 is one of the five isoforms of genes encoding heparan sulphate biosynthesizing enzymes, heparan sulphate sulphotransferases (HS3STs). Heparan sulphate binds to specific proteins such as antithrombin and several growth factors, and thereby regulates various biological processes including anticoagulation and angiogenesis (Rosenberg et al. 1997). HS3STs catalyze sulfation of monosaccharide sequences of heparan sulphate, which is believed to be critical for binding to the target proteins.HS3ST3B1 has a closely related isoform, HS3ST3A1, which also has similar patterns of tissue expression and encodes a protein with similar enzymatic activity. Together with the nature of this type of catalytic enzyme, wherein changes in dosage usually do not affect the system, existence of a paralog with similar enzymatic properties suggest that duplication or deletion of one allele ofHS3ST3B1 may not affect heparan sulphate biosynthesis.
Tektin includes a family of proteins and represents one of the components of motile and primary cilia associating with the major structural component of cilia, microtubules (Linck and Langevin 1982;Linck et al. 1985; Steffen and Linck 1988). Tektins have been best studied in sea urchins, a species in which three isoforms have been isolated; tektin A1, tektin B1, and tektin C1. Mammalian homologs for tektin B1 and tektin C1 have been isolated (GenBank accession no. NM_014466, NM_011902 and NM_011569) (Norrander et al. 1998; Iguchi et al. 1999). In the CMT1A/HNPP region, we identifiedTEKT3 as the first homolog for tektin A1 in mammals. Like other tektin homologs, it is preferentially expressed in testis. Tektin A1 and tektin B1 are thought to be assembled as heterodimers to comprise the tektin filament, and interact with tubulins to form the basis of the high degree of stability of doublet microtubules (Pirner and Linck 1994). In the mouse sperm, the tektin B1 homologous protein tekt2 is localized in flagella, strongly suggesting that tektins may play essential roles in formation of sperm and in sperm motility (Iguchi et al. 1999). Loss of TEKT3 may reduce the motility of the sperm of HNPP patients because of their haploid nature.
Relevance to CMT1A/HNPP Genomic Disorders
Of the new LCRs found in the CMT1A/NHPP region, LCRA2 and LCRB are present in a tandem orientation and flank PMP22, suggesting that they have the potential to be substrates for unequal homologous recombination leading to duplication or deletion of PMP22. Four families with alternate size duplication or deletion were reported previously (Ionasescu et al. 1993; Palau et al. 1993; Valentijn et al. 1993; Chapon et al. 1996). Genetic studies with a few markers showed that the proximal break points of these cases are located close to or within the proximal CMT1A–REP, and the distal break points mapped between PMP22 and D17S125 (Ionasescu et al. 1993;Palau et al. 1993; Valentijn et al. 1993; Chapon et al. 1996). Therefore, at least in these cases, recombination between the LCRs found in this study are unlikely to be involved in the small duplication or deletion. Additional analyses for LCR in this region failed to identify any significant stretches of homologous sequence (>1 kb) that may serve as substrates for such alternative homologous recombination events.
Most of the genes identified in this study revealed extremely low expression in adult tissues but obvious expression in fetal tissues. It is surprising that these embryonic genes have no developmental effect on the individuals with duplication or deletion of the 1.4-Mb region. The observation that to date PMP22 is the only gene responsible for CMT1A/HNPP due to the mechanism of gene dosage accompanied by duplication or deletion of this region suggests that dosage sensitivity may be a unique property of PMP22 but not of the other genes in the 1.4-Mb region. The sequence of most of these genes contains insufficient information to estimate their function. However, the cumulative data suggest that only 1 in 21 genes, at least in this portion of the human genome, is sensitive to dosage effects.
Evolution of New Genes, HREP and CDRT1, by DNA Rearrangement
Identification of the COX10 gene spanning the distal CMT1A–REP and only one exon (pseudoexon VI) in the proximal CMT1A–REP indicates that the distal copy is the original and the proximal CMT1A–REP represents a duplicated copy (Murakami et al. 1997a; Reiter et al. 1997). Evolutionary studies reveal that this insertional event occurred between gorilla and chimpanzee (Kiyosawa and Chance 1996; Reiter et al. 1997; Boerkoel et al. 1999; Keller et al. 1999). Subsequently, another gene, HREP, was identified close to the proximal CMT1A–REP (Kennerson et al. 1997, 1998). HREPis transcribed toward the telomere from outside the proximal CMT1A–REP and terminates within the proximal CMT1A–REP. The last exon ofHREP occurs at the same position, but on the complementary strand of COX10 pseudoexon VI (Kennerson et al. 1997).
Interestingly, we found that a mouse gene homologous to humanHREP does not share the region after exon V with humanHREP, but instead matches CDRT1, which is adjacent to the proximal CMT1A–REP on the telomeric side. Therefore,CDRT1 and HREP are likely to be parts of anAncestral Gene before theIntegration of Proximal CMT1A–REP (AGIP) (Fig. 6). The CMT1A–REP insertional event, which is estimated to have occurred during primate speciation between gorilla and chimpanzee, divided AGIP into two genes, HREP andCDRT1. These findings show an example of evolution of new genes by DNA rearrangement during mammalian genome evolution. The first half of AGIP became HREP utilizing a part of CMT1A–REP as a new terminating exon, whereas the last exon ofAGIP became a single exon gene CDRT1. Interestingly, expression profiles of these two genes are different; HREP is expressed in heart and skeletal muscle, whereas the major expression ofCDRT1 is observed in pancreas. Furthermore, a region inAGIP between the HREP syntenic portion andCDRT1 syntenic portion was likely to be lost during the CMT1A–REP integration, suggesting that this insertional genomic rearrangement was accompanied by loss of a genomic fragment. Further evolutionary analysis of the genomic region surrounding proximal CMT1A–REP in chimpanzee and gorilla may elucidate the mechanism of integration of the CMT1A–REP.
In conclusion, we have evaluated the 1.4-Mb finished genomic sequence of the CMT1A/HNPP region. Data obtained from this genome-sequencing study enable new insights into human genome architecture and mammalian genome evolution, show evolution of new genes by genome rearrangements during primate speciation, and add to the plethora of information being created by the complete nucleotide sequencing of the human genome.
METHODS
Construction of Physical Maps of the 1.4-Mb CMT1A/HNPP Region
We implemented two independent approaches to construct the physical map of the CMT1A/HNPP genomic region. The first approach utilized STS content-based mapping performed at Baylor College of Medicine. We used the end sequences of the multiple cosmid clones from a previously constructed cosmid contig of this region (Murakami and Lupski 1996) to screen PAC (P1 artificial chromosome; RPCI-1 Rosewell Park Cancer Institute, Buffalo, NY) and BAC (bacterial artificial chromosome; CITB California Institute for Technology) libraries by PCR on DNA pools and/or by filter hybridization. Eight known genetic markers and the PMP22 gene were also used as probes. Overlaps of each large insert genomic clone were evaluated byEcoRI fingerprinting by use of a FluorImager (Molecular Dynamics), as described elsewhere (Marra et al. 1997).
A parallel and alternative approach used YAC-based mapping conducted at the Whitehead Institute Center for Genome Research as a part of the effort to sequence the entire human chromosome 17. To create reliable physical maps despite significant amounts of low-copy repetitive sequence, we used a high density of unique markers. In addition to pre-existing markers, new markers were generated from shotgun sequences derived from pulsed-field gel-purified YACs. Overlapping YACs from the CEPH Mega-YAC library (Chumakov et al. 1995) that were not known to be chimeric based on STS content (Hudson et al. 1995) were selected from the CMT1A region (Pentao et al. 1992). Each YAC was fractionated and subcloned separately into M13. Single-sequencing reactions were performed on several hundred subclones from each YAC and the resulting sequences contained from 20%–60% yeast DNA, depending on the YAC. Thirty-eight base pair overgos were designed (Ross et al. 1999) and further tested by hybridization to eliminate probes that contained highly or moderately repetitive sequences that escaped detection during their design. BAC library (RPCI-11) screening was by hybridization with pools of up to 40 overgos derived from a single YAC, with an average density of 30 overgos per Mb of genomic region. Positive clones from the library screen were streaked on agar plates to obtain single colonies and one clone from each positive address was rearrayed into new 96-well plates. To generate marker content maps, replica filters made from the 96-well plates were hybridized individually with each of the overgos used in the library screen, as well as overgos derived from overlapping YACs, and overgos representing other markers mapped in the region. Markers that hybridized to greater than the expected number of clones were not included in the final map, nor were markers that were not linked by at least two clones. Clones that did not share at least two markers with an overlapping clone were not included in the map. The final density of markers in the BAC map of the region was ∼1 marker every 10 kb. This high-density physical mapping generated an overlapping contig with 8- to 10-fold coverage. Combining these two physical maps, clones with a minimal tiling path were selected for sequencing (Fig. 1).
Shotgun Library Construction, DNA Sequencing, and Sequence Data Analyses
Subclone libraries were constructed for each human genome containing bacterial clone and shotgun sequencing, assembly and finishing was performed as described (International Human Genome Sequencing Consortium 2001). A single annotated gap remains in the sequence of RP11–726O12 (AC005517). PCR amplification of template DNA from the corresponding large-insert genomic clone followed by sequencing revealed that the gap contains 439 bp with an extremely high content of GA repeat. The repeat content is probably responsible for the difficulties encountered in cloning and sequencing this gap region. The sequence from each BAC/PAC clone was assembled into a larger sequence contig by use of Sequencher (Gene Codes). These data were analyzed by the NIX analysis program (Nucleotide Identification of unknown sequences, UK MRC Human Genome Mapping Project; http://www.hgmp.mrc.ac.uk), a Web-based package of gene analysis software (including GRAIL, Fex, Hexon, MZEF, Genemark, Genefinder, FGene, BLAST, Polyah, RepeatMasker andTRNAscan). Each region that contained a potential gene was individually analyzed by additional gene prediction and protein analysis programs, by use of the ExPASy proteomics server (Expert Protein Analysis System; http://www.expasy.ch). Putative core promoter and transcription-binding sites were analyzed by TESS (http://www.cbil.upenn.edu/tess/index.html),Human Core-Promoter Finder(http://sciclio.cshl.org/genefinder/CPROMOTER/human.htm),TSSG, and TSSW (BCM GeneFinder;http://dot.imgen.bcm.tmc.edu:9331/gene-finder/gf.html).RepeatMasker was independently run to identify interspersed repeat sequences. A genetic map of chromosome 17 with raw data from polymorphic genetic markers within this region was obtained from the Marshfield Web site (http://www.marshmed.org/genetics) to evaluate genetic/physical map correlations (Broman et al. 1998).
Northern Blotting and RT–PCR Analyses
Expression profiles and the size of each transcript was determined by multiple tissue Northern blotting (Clontech). Primers from the unique 3′ untranslated region of each isolated gene were designed by use of web-based software, Primer3(http://www-genome.wi.mit.edu/genome_software/other/primer3.html). Corresponding BAC/PAC clones were used as template DNA for PCR to generate probes to minimize the chance of amplification of gene family members and pseudogenes mapping elsewhere in the genome. RT–PCR was performed for some of the predicted genes by use of first-strand cDNA from various adult and fetal tissues (Clontech).
Acknowledgments
We thank Yi-Mieng Chang, Thearith Koeuth, and Stephen Ansley (Baylor College of Medicine) for their technical assistance. We also thank Will FitzHugh, George Grant, Rob Nahf, Diane Gilbert, and Boris Pavlin for their technical support of the WIBR mapping activities and all members of the WI/MIT Center for Genome Research Sequencing Group. K.I. and L.T.R. are supported by postdoctoral fellowships from the Charcot-Marie-Tooth Association. This research was supported in part by grants from the National Human Genome Research Institute to E.S.L., the National Eye Institute to N.K. (R01 EY12666), and the National Institute for Neurological Disorders and Stroke (R01 NS27042) and the Muscular Dystrophy Association to J.R.L..
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Present address: Department of Biology, University of California San Diego, La Jolla, CA 92093, USA.
-
↵5 Corresponding author.
-
E-MAIL jlupski{at}bcm.tmc.edu; FAX (713) 798-5073.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.180401.
-
- Received January 18, 2001.
- Accepted February 27, 2001.
- Cold Spring Harbor Laboratory Press








