
Envelope-Class Retrovirus-Like Elements Are Widespread, Transcribed and Spliced, and Insertionally Polymorphic in Plants
Abstract
Retrotransposons and retroviruses share similar intracellular life cycles and major encoded proteins, but retrotransposons lack the envelope (env) critical for infectivity. Retrotransposons are ubiquitous and abundant in plants and active retroviruses are known in animals. Although a few env-containing retroelements,gypsy-like Athila, Cyclops, andCalypso and copia-like SIRE-1, have been identified in plants, the general presence and functionality of the domain remains unclear. We show here that env-class elements are present throughout the flowering plants and are widely transcribed. Within the grasses, we show the transcription of the envdomain itself for Bagy-2 and related retrotransposons, all members of the Athila group. Furthermore, Bagy-2transcripts undergo splicing to generate a subgenomic envproduct as do those of retroviruses. Transcription and the polymorphism of their insertion sites in closely related barley cultivars suggests that at least some are propagationally active. The putative ENV polypeptides of Bagy-2 and rice Rigy-2 contain predicted leucine zipper and transmembrane domains typical of retroviral ENVs. These findings raise the prospect of active retroviral agents among the plants.
[The sequence data described in this paper have been deposited as follows: Bagy-2 elements, EMBL accession nos.AF254799 and AJ279072; an alignment of 328 gypsy-likert sequences, accession no. DS44537; barley envsequences, accession nos. AJ298028–AJ298032; cDNA sequences for the spliced env subgenomic RNAs, accession nos.AJ311200–AJ311202; genomic sequences for env-classrt, accession nos. AJ295085–AJ295111; cDNA sequences forenv-class rt, accession nos. AJ295112–AJ295139; representatives of polymorphic Bagy-2 bands from IRAP gels, accession nos. AF363958, AF 363959, and AY029538.]
Retrotransposons and retroviruses are related genetic elements replicating through a cycle of successive transcription, reverse transcription, and integration into the genome. Retroviruses differ from retrotransposons in their being infective. The infectivity of mammalian retroviruses depends critically on their encoded envelope (ENV) glycoproteins (Frankel and Young 1998), which recognize receptor proteins on the surface of host cells, allowing adsorption to them, and help to mediate subsequent penetration of the plasma membrane.
Retroviruses are more similar to one particular class of plant, fungal, and invertebrate retrotransposons, those resembling the type-elementgypsy of Drosophila melanogaster (Kumar and Bennetzen 1999). The encoded capsid protein (GAG), proteinase (PR), integrase (IN), reverse transcriptase (RT), and RNaseH (RH), bounded by long terminal repeats (LTRs), are organized 5′-LTR-GAG-PR-RT-RH-IN-LTR-3′ in gypsy-like retrotransposons and retroviruses,env being found in retroviruses between in and the 3′ LTR. The other major group, the copia-like retrotransposons, are organized 5′-LTR-GAG-PR-IN-RT-RH-LTR-3′. The strong internal sequence similarities, respectively, in thecopia-like and gypsy-like groups suggest that they are lineages that have been separated since early in eukaryote evolution (Xiong and Eickbush 1990). Some invertebrate retrotransposons, including gypsy from Drosophila, which is infective, contain env domains (Song et al. 1997;Malik et al. 2000). These have therefore been classified as errantiviruses (Boeke et al. 1999).
The restrictions imposed by plant cell walls to membrane–membrane interactions might suggest that envelopes and ENV glycoproteins would not be as useful to plant viruses as to animal viruses, explaining the lack of reported plant retroviruses. However, some plantgypsy-like retroelements have been shown to contain domains reminiscent of animal env, the Athila/Tat1clade of Arabidopsis thaliana (Pélissier et al. 1995; Wright and Voytas 1998) and the related legume elements Cyclops of pea and Calypso of soybean (Chavanne et al. 1998;Peterson-Burch et al. 2000). A unique copia-like,env-containing element, SIRE-1 has also been described for soybean (Laten et al. 1998).
These findings suggest that either the plant env domains have been acquired independently by transduction in scattered instances or they are common and have been passed by descent to a wide group of plants. Phylogenetic analyses strongly suggest that the insect errantiviruses transduced an env gene from a baculoviral source (Malik et al. 2000; Rohrmann and Karplus 2001), but the same analyses left the origin of the scattered plant env domains open. We have investigated this question and show here thatenv-class retroelements are present throughout the flowering plants and are widely transcribed. Analysis of the gypsy-likeBagy-2 retrotransposons of the Athila clade shows that their env domains in particular are also transcribed widely and, furthermore, spliced in a manner similar to that of retroviruses. Bagy-2 is polymorphic in its insertion sites in closely related cultivars, suggesting active retrotransposition. The predicted polypeptides of Bagy-2 and clade memberRigy-2 of rice contain the basic features of ENV.
RESULTS
Bagy-2 and Rigy-2 Retrotransposons Contain anenv Domain
We recently identified a new retrotransposon, Bagy-2, in barley (Shirasu et al. 2000). Sequences from two Bagy-2 clones (AF254799 and AJ279072) reveal that the element is ∼10 kb overall, containing LTRs of 1520–1537 bp and an internal protein-coding region organized similarly to other gypsy-like elements. These LTRs are relatively long for plant retrotransposons, as are those of another barley retrotransposon, BARE-1 (Manninen and Schulman 1993;Suoniemi et al. 1997). Immediately interior to the 5′ LTR is a putative primer binding site (PBS), which is identical in 17 of 18 nucleotides at the 3′ end of a tRNA-Glu from Schizosaccharomyces pombe(Wong et al. 1979). The use of tRNA-Glu is fairly unusual among plant retrotransposons, but is shared with Cyclops-2 (Chavanne et al. 1998). A possible polypurine tract (PPT) was observed 1 base upstream of the 3′ Bagy-2 LTR sequence, which is identical to that of Athila and again very similar to Cyclops-2(12 of 14 bases), both gypsy-like elements. The derived amino-acid sequence of the Bagy-2 internal domain, encompassing GAG, proteinase, reverse transcriptase, RNase H, and integrase domains, are also most similar to those of Athilaand Cyclops-2, and gypsy-like in domain order.
The Rice Genome Research Program has made it possible for us to identify Bagy-2 homologs in rice, which we nameRigy-2 in parallel with the name for the barley element. Four copies are present in Genbank (Release 125.0, August 15, 2001), all four containing other retrotransposons nested within as follows:AP003054.2 (nucleotides 36243–53386), AP003208.2 (nucleotides 50267–65711, reverse), AP003414.3 (nucleotides 115228–120398, partial), and AC0022352.5 (nucleotides 13684–113587). The consensus element contains LTRs of 1171 bp and a total size of 9753 bp.
In the 3′ end of the internal domain, Bagy-2 andRigy-2 contain a region the position and structure (see below) of which match env. To establish that this domain is a general feature of Bagy-2 retroelements, we designed flanking primers. Amplification reactions with these primers and barley genomic DNA or barley BAC clones as the template always yielded the 2.4-kb band expected if the putative env domain were present (not shown). Sequences from this domain of two cloned elements, Bagy-2-1 and Bagy-2-2 (AF254799 and AJ279072) are 96% identical on the DNA level and resemble env (see below).
Bagy-2 Elements Containing env Are Transcribed In Barley and Widespread In Grasses
The barley sequences enabled us to design primers within theBagy-2 env in order to examine plants other than barley for the presence of an env domain. These produced products, shown in Figure 1A, similar in size to one another from across the grass family. We used the same primers (p3 and p4 in Fig. 2A) to investigate whetherBagy-2 env is transcribed. Amplification reactions yielded products, identical in length to those from genomic DNA, from barley RNA of various tissues (Fig. 1B). Control reactions lacking mRNA or the reverse transcription step failed to yield products, confirming the transcriptional origin of the amplified products. Four amplified cDNAs, respectively, from leaf, cell culture, pollen, and embryo were cloned and sequenced (AJ298029–AJ298032). The predicted proteins of these cDNAs were 86.5% similar to each other. They are also 81% similar to a barley leaf EST (BE437584), providing support for transcription of Bagy-2 elements.
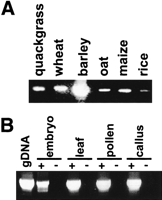
Detection of genomic and expressed env domains. (A) PCR amplification of a 385-bp segment of env domains from 10 ng of genomic DNA of quackgrass (Elymus repens), wheat (Triticum aestivum) barley (Hordeum vulgare), oat (Avena sativa), maize (Zea mays), and rice (Oryza sativa). Bands of identical size could be detected in other Hordeum spp. as well as in rye (Secale cereale). Species other than wheat and quackgrass derive fromPoaceae subfamilies different from that of barley. (B) RT–PCR amplification from RNA of various barley tissues. As a standard, PCR with 10 ng of genomic DNA is shown. The reactions for lanes labeled + contained a reverse transcription step to convert the RNA to cDNA; those labeled − lacked nucleotides at this step as controls for genomic DNA contamination.
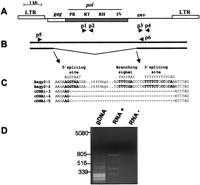
Splicing of Bagy-2 RNAs. (A) Diagram of theBagy-2 genome showing placement of the gag,pol, and env coding regions, and the products encoded. Primer pairs used for amplifying the rt andenv domains are shown, respectively, as p1, p2 and p3, p4. (B) Diagram of the full-length (top) and spliced (bottom) RNAs together with the primer pair (p5, p6) used to detect the spliced product. (C) Sequences of the twoBagy-2 genomic clones and three leaf cDNA clones at the splice site. Consensus splicing signals are shown above. Consensus nucleotides are indicated in bold. (D) Agarose gel analysis of the RT–PCR amplification products using primers p5 and p6. The lanes display reactions containing gDNA, genomic DNA; RNA+, with template leaf RNA; RNA−, a control lacking the nucleotides required for reverse transcription.
The Bagy-2 env Domain Is Expressed From Spliced mRNAs
In the retroviruses, ENV proteins are translated from a spliced subgenomic mRNA, which, through the splicing process, lacks the genes for GAG and the POL products (PR, IN, RT, RH) in the mature transcript (Vogt 1997). To examine whether the Bagy-2 env transcripts were being spliced in a similar manner, we carried out amplification reactions using one primer in the LTR and another inside theenv domain (pair p5, p6), as diagramed in Figure 2A. This primer pair amplifies from RNA an ∼800-bp fragment (Fig. 2D), which is not present in genomic DNA or in the control reaction and differs from the products in Figure 1, which were amplified with internalenv primers (Fig. 2A, p3, p4). The subgenomic RNA is therefore not a product of an internally deleted Bagy-2 present as such in the genome. No unspliced, full-length transcripts were detected by use of these primers, perhaps due to both the efficiency of the splicing reaction in vivo and the comparative inefficiency of both long cDNA generation and long PCR amplification in vitro.
The 800-bp product from cDNA of barley leaf tissue was cloned and sequenced (AJ311200–AJ311202); analysis indicated that it is likely to correspond to a spliced mRNA. The first 826 bp of the fragment matches the Bagy-2 LTR, PBS, untranslated leader, and the beginning ofgag (Fig. 2B). The remainder of the sequence is discontinuous with gag, but matches instead the 146 bp of the envregion upstream of the p6 primer used in the reactions (Fig. 2B). This correspondence with the segment of the env region expected from the primer placement indicates that the product is not due to priming at a secondary site in the amplification reaction. The putativeenv splice site is furthermore identical in three separate cDNA clones (Fig. 2C) and the flanking dinucleotide matches the consensus GT/AG of intron splice sites (Brown et al. 1996; Rogozin and Milanesi 1997). Moreover, the putative donor and acceptor splicing signals in Bagy-2 are very similar to the consensus plant splicing signals (Fig. 2C; Brown et al. 1996; Rogozin and Milanesi 1997). The consensus branching signal, required for the splicing reaction (Brown et al. 1996), is also conserved, with the exception of the terminal pyrimidine (Fig. 2C).
We have cloned and sequenced (data not shown) the prominent ∼400-bp and ∼300-bp PCR products (Fig. 2D). They resulted not from alternative splicing, but corresponded to deleted forms of theBagy-2 genomic transcripts that lack the gag domain, but have the final terminal region of the integrase and of theenv domain at least until the primer. The intensity of these products may not reflect the prevalence or transcriptional strength of deleted or nested Bagy-2 derivatives, but rather the more efficient amplification of shorter products. The demonstration of subgenomic mRNAs, originating in the LTR, also offers support forBagy-2 transcription being element promoted rather than read-through by cellular promoters into retroelement fragments.
The Predicted Bagy-2 and Rigy-2 ENV Has Key Conserved Domains
We examined the predicted Bagy-2 ENV sequences for diagnostic motifs found in ENVs. In retroviruses, the ENVs are glycosylated at Asn residues. As diagramed in Figure3A, the predicted ∼47-kD ENV ofBagy-2 has at least two consensus N-glycosylation sites. Leucine zippers (Cohen and Parry 1986) in the transmembrane domain of ENVs mediate membrane fusion, an essential step in the infective stage of the retroviral life cycle (Chen et al. 1998). In the Bagy-2ENV, a well-defined leucine zipper is predicted in the position expected from retroviral ENVs (Fig. 3A). A structural prediction algorithm, effective for such domains (Rost et al. 1995), strongly predicted the presence of hydrophobic, membrane-spanning helices in the putative Bagy-2 translation (Fig. 3A), as expected for ENVs. The Rigy-2 copies in GenBank, including the envregions, contain frameshifts and stop codons. The predicted consensus for the Rigy-2 ENV (Fig. 3B) contains seven putative N-glycosylation sites, one putative cleavage site, and a leucine zipper (one of the Leu has been substituted by Ile). It also has two putative transmembrane domains, although the second one is predicted by theSOUSI program, but it is not predicted clearly byTMHMM, which was used for the probability plot in Fig. 3B.
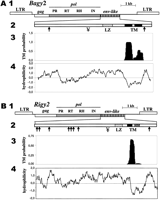
Features of the Bagy-2 and Rigy-2 retrovirus-like retrotransposons and their predicted ENV products. (A)Bagy-2. (1) Diagram of the element drawn to scale. Between the long terminal repeats (LTRs) are the predicted gagdomain encoding the capsid protein, the pol domain encoding the proteinase (PR), reverse transcriptase (RT), RNaseH (RH), and integrase (IN), and the putative envelope (env) domain. (2) Diagram of the predicted ENV protein showing putative N-glycosylation sites (↓), consensus proteinase cleavage site (¥), leucine zipper (LZ), and transmembrane domain (TM). (3) Probability plot for occurrence of transmembrane domains. (4) Hydrophilicity plot for the predicted ENV protein. (B)Rigy-2. Features of Rigy-2 as for Bagy-2 inA.
Insertional Polymorphisms Indicate Bagy-2 Integrational Activity In Barley
The structural conservation and transcriptional activity of theBagy-2 elements suggest that they may be integrationally active. We looked for signs of transpositional activity as polymorphisms for sites of Bagy-2 integration. For this purpose, we applied the IRAP (inter-retrotransposon amplified polymorphism) technique (Kalendar et al. 1999), which uses outward-facing primers matching retrotransposon LTRs to amplify genomic regions lying between two retroelements. Variations in the retroelement insertion pattern between samples are identified as polymorphisms for the presence of bands in the resolved PCR reaction products and imply retrotransposon insertion events since the last common ancestor of the genotypes tested.
We chose a set of 29 European barley varieties well characterized with molecular markers (Ellis et al. 1997; Russell et al. 1997) and applied IRAP. The levels of polymorphism, as displayed in Figure4, seen with IRAP for Bagy-2 were at least as high as for BARE-1 (Waugh et al. 1997; Kalendar et al. 1999), a retrotransposon known to be a dynamic component of the barley genome (Jääskeläinen et al. 1999; Vicient et al. 1999). The European germplasm pool of elite malting varieties represented in the sample material is fairly narrow, and is also derived from a limited founder population brought with the spread of agriculture into Europe. Detectible polymorphisms for retrotransposon integration thus suggest recent mobility on a scale of decades to millennia.
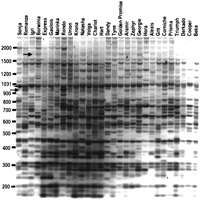
Polymorphism at Bagy-2 integration sites in a set of European barley cultivars. The PCR products were generated from template DNA for each cultivar (top) with the IRAP (inter-retrotransposon amplified polymorphism) technique (Kalendar et al. 1999). The products have been stained with ethidium bromide during agarose gel electrophoresis; the gel is shown as a negative image. Arrows indicate bands cloned for sequence analysis. The size markers (left) are in basepairs.
We sought to confirm that the products detected from theBagy-2 IRAP reactions were specific for particular loci in the genome by cloning and sequencing polymorphic bands of 1598 bp, 998 bp, and 950 bp (arrows, Fig. 4; representatives of each size, accession nos. AF363958, AF363959, and AY029538). Respectively, one, two, and two products of these size classes from different barley cultivars were cloned from IRAP gels and sequenced. For each sequence, the LTR termini were present on the flanks of the insertions as expected. For the pairs of sequences of identical size, the genomic sequences intervening between the Bagy-2 insertion sites were virtually identical. This indicates that IRAP based on Bagy-2is specific and reproducible and has sufficient resolution to differentiate insertions at particular loci.
Env-Class, Athila-Like Elements Are Ubiquitous In Flowering Plants
Taken together, the foregoing results indicate thatenv-containing elements are probably ubiquitous and active in the grasses. However, we could not directly amplify envdomains from species outside of the grasses with the primers effective for the grasses. Because the heterogeneity of env (Lerat and Capy 1999; Malik et al. 2000) may preclude the design of universal primers, we chose another route to examine whether env-class elements are widespread in the plants. An alignment of 328gypsy-like rt sequences from known retrotransposons and database accessions of all organisms was constructed (DS44537) on the basis of earlier alignments (Xiong and Eickbush 1990).
The resulting phylogenetic tree based on these sequences and subsets thereof (Fig. 5) is consistent with one that was reported recently (Marín and Lloréns 2000). The plantgypsy-like elements were resolved into two lineages, one universally lacking env domains and the other containing the sequences we amplified together with the other elements containingenv-like or long 3′ regions (the two boxed groups in Fig. 5). The latter set was further divided into two clear groups, one containing Tat4, RIRE2, Grande1, RetroSor, the Tat branch, and theAthila branch including Athila, Calypso-2, Calypso-1,Cyclops-2, Diaspora, Tfcl1, Bagy-2, Rigy-2and all of the amplified elements. The Tat group has LTRs <550 bp and 3′ noncoding regions >2 kb, whereas the Athilagroup has LTRs >1.2 kb and overall lengths >11 kb. TheAthila clade, containing Bagy-2, furthermore contains all other sequenced gypsy-like plant elements having a putative env domain. Our alignment indicated that theAthila-like rt sequences are distinct enough from the others to permit building of consensus primers to amplifyenv-element-specific rt domains (p1,p2).
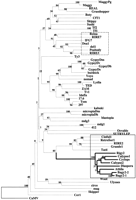
Phylogenetic analysis of the reverse transcriptase (RT) domain sequences of Ty3/gypsy-class retrotransposons. The neighbor-joining tree was inferred from Kimura estimates of sequence distance. Numbers in the branches are the bootstrap values from 500 replicates. Plant elements with env or long 3′ domains are boxed by a solid line and the Athila-clade lines are in bold; gypsy-like plant elements lacking env are boxed by a broken line. A complete list of accession numbers and details for the elements is available at the EMBL European Bioinformatics Institute (EBI) public alignments Web site (http://www3.ebi.ac.uk/Services/align/listali.html) under the accession no. DS44537.
First, we tested these primers with barley genomic DNA and obtained the expected band of ∼390 bp. Amplifications were then carried out with genomic DNAs of species representing the diversity of land plants (Suoniemi et al. 1998). The angiosperms examined all showed, as in Figure 6, a single band of the expected size, whereas nonangiosperms failed to yield a product. To legitimate the specificity of the amplifications, 27 rt sequences were determined for 7 species. These were incorporated into an alignment ofrt domains from plant retroelements, analyzed on the basis of the predicted translations, and a neighbor-joining tree was constructed. All 27 clones fell into the Calypso-Athila-Bagy-2clade, which has a 100% bootstrap value, which is shown in Figure 5.

Detection of env-class retrotransposons by rt domain in genomic DNA of diverse angiosperms. A 390-bp domain specific to the group of gypsy-like retrotransposons containing envwas amplified by PCR using primers p1 and p2 and resolved by agarose gel electrophoresis. The samples consisted of the following: (a) Arabidopsis thaliana (Columbia); (b)Brassica olearecea (var. botrytus); (c) Citrus sinensus; (d) Betula sp.; (e)Pisum sativum; (f) Dianthus sp.; (g) Nicotiana tabacum (SR1); (h)Anenome nemorosa; (i) Secale cereale; (j) Triticum aestivum; (k) Hordeum vulgare; (l) Avena sativa; (m)Oryza sativa; (n) Echinodrus muricatus; (o) Nymphaea carerulea. The phylogenetic relationships between most of these species has been displayed earlier (Suoniemi et al. 1998). All angiosperms tested (not all shown) were positive; all tested older plant groups including a fern (Neprolepsis exaltata), Psilotum, a cycad (Cycas circinalis), and conifers in the Pinaceae were negative.
Given the specificity of the rt primers for env-class elements, we examined transcriptional activity of this group by RT–PCR for the rt domain using RNAs from five species, as seen in Figure 7. Transcripts equivalent in size to those from genomic DNA were detected in various barley tissues and in all tested monocots and dicots. The transcriptional activity in barley is consistent with that observed for the env domain directly (Fig. 1). A total of 27 rt cDNAs were sequenced. The high similarity of these sequences to two cDNA accessions (AB007466 andAB007467) from the guard cells of bean Vicia faba as well as to unannotated EST clones from wheat (BE424901), sorghum (AW672403), the legume Medicago trunculata (AW585806), and soybean (AW278189) confirms that the env-class of gypsy-like retroelements are broadly expressed in plants.
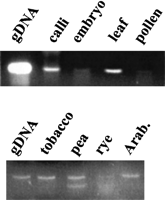
Transcription of env-class retrotransposons. Transcripts were detected by RT–PCR amplification of the rt domain using primers p1 and p2 and produced a product identical to that in Fig. 6. (top panel) Transcription in various barley tissues. Controls consisted of genomic DNA (gDNA in figure) and reverse transcription in the absence of nucleotides (no band produced, not shown). Bands from embryo and pollen are both weak but present. (bottom panel) Transcription in the leaves of other species, the upper band being identical in size to that in barley. Giant radican (Echinodorus muricatus, not shown) also produced a band of identical size.
We aligned the predicted translations of 43 genomic or cDNA sequences from barley and 16 from other plants with those of database rtsequences. The neighbor-joining tree (Fig.8) constructed from this alignment clearly separates (80% bootstrap value) the sequences into two large clades, one containing only cereal accessions and the other containing both cereal and dicot members. The cDNA accessions are not resolved from those deriving from genomic DNA; this implies that the actively transcribed elements are not a subgroup. The mixed clade is in turn divided into two groups (62% bootstrap value), one containing 4 monocots of the 10 accessions and the other only 2 monocots of 20 accessions. These data suggest that an env-class subfamily grew in prevalence following the divergence of the evolutionary line leading to the cereals from its common ancestor with the dicots. All of our sequences, together with the env-class plant retroelements in the database, are completely (100% bootstrap value) separated from nonplant elements, both with and without env, as well as from plant gypsy-like elements without env (Fig. 5).

Phylogenetic analysis of the reverse transcriptase (rt) domain sequences of Bagy-2 and related Ty3/gypsy retrotransposons. The neighbor-joining tree was inferred from Kimura estimates of sequence distance. Numbers at the nodes are the bootstrap values of 500 replicates. Some of the sequences were obtained from GenBank (see Fig.5 for accessions); the others, determined by us (accession nos.AJ295085–AJ295139), are in capital letters. Genomic sequences are labeled with the suffix -G, cDNA sequences from leaves, -L; cDNA sequences from cultured cells, -C.
DISCUSSION
The data here establish that a distinct class ofgypsy-like, env-class retrotransposons related toAthila is widespread and transcribed in flowering plants (angiosperms). Barley Bagy-2 and rice Rigy-2, as well as all other sequenced members of this group, encode a predicted ENV with conserved features. Bagy-2 is transcribed in all tissues examined, is spliced by conserved signals as is universally found for retrovirus env, and displays insertional polymorphism in closely related barley cultivars, implying recent transpositional activity. In other cereals, plants sufficiently close to barley that the env primers may be used directly, envtranscription was also directly demonstrated.
The ENV is typically encoded by a subgenomic spliced transcript specifying a protein cleaved by host proteases into the glycosylated surface and the transmembrane polypeptides of the infectious virus. The ENV sequences constitute a very heterogeneous collection and only very closely related sequences reveal extensive recognizable similarities in primary structure (Lerat and Capy 1999). Despite the considerable size and sequence diversity among retroviral envelope proteins, some regions of similarity distributed throughout the sequence can be found. This sequence diversity dictated a different approach to demonstrating the ubiquity of env-containing, gypsy-like elements in the plants, one that relied on their otherwise being similar and conserved in sequence, in particular, in the rt domain. Using primers specific for such env-class elements, we were able to show their presence in all angiosperm genomes examined.
The Bagy-2 and other gypsy-likeenv-containing elements appear to have been active and propagationally successful. Env-class elements are pervasive throughout plants separated by tens of millions of years of evolution. Of the retroelements, the env-class Athila is one of the most abundant in Arabidopsis thaliana (Pélissier et al. 1995). Bagy-2 appears to be approximately as abundant in barley, present in excess of 104 copies (C. Vicient, unpubl.), as the BARE-1 elements, an activecopia-like element in barley and other cereals (Vicient et al. 2001). Another member of the Athila clade, Cyclops, is found in 5000 copies in the genome of Pisum sativum(Chavanne et al. 1998).
Furthermore, several considerations suggest that these envdomains have evolved and have been maintained under functional constraint. The env domain is found in multiple distinct element families, which are, however, united on a strongly supported clade, and within these families it is relatively conserved. For example, between Athila and Athila1-1, the type elements of the group including Bagy-2, the env-like ORF shares ∼34% similarity with >400 residues. Moreover, these coding domains for putative ENVs encode transmembrane domains, the most universal feature of retrovirus and animal errantivirus envelope proteins, suggesting that they could interact with the envelope of a virus-like particle. Athila and the closely related retrotransposon Athila2-1 also encode a transmembrane domain near the N terminus of the ORF at a position typically occupied by the secretory signal sequences in envelope proteins. Putative glycosylation sites and endopeptidase cleavage domains, both typically found in mammalian retroviruses and animal errantiviruses, can also be identified in the env-like genes of this group of gypsy-like retrotransposons from plants.
The identification of spliced Bagy-2 RNA provides further evidence of the striking conservation of retrovirus-like aspects ofenv-class retrotransposons. Our data suggest that regulation at the level of RNA splicing may be a factor contributing substantially to Bagy-2 expression. It is the full-length RNA that serves as the template for synthesis of the GAG structural protein and the POL synthetic proteins including PR, IN, RT, and RH. The ratio of full-length to ENV-coding transcript may be controlled by the splicing efficiency, which, in turn, would be regulated by the host splicing factors.
Given that at least some Bagy-2 and other retrotransposons coding for ENV are active, the question of the function of this glycoprotein in the retrotransposon life cycle becomes sharper, yet the answer remains uncertain. An ENV protein is clearly not needed for agypsy-like or copia-like retrotransposon to become abundant. Nevertheless, the capacity through ENV-mediated budding and infection to move extracellularly, or from individual to individual, may offer selective advantages through increased replication potential. Infection and horizontal transfer, for a genetic entity such as a transposable element that can integrate into a genome, are similar phenomena. The phylogenetic distinctness of theAthila clade might be expected if horizontal transmission spread these elements between species. Alternatively, plant ENVs may have intracellular or intraplant rather than infective roles, either in element replication or in cell-to-cell movement within the plant.
A replication or infection-competent plant errantivirus must be identified, its virus-like particle visualized, and its life cycle characterized in order to resolve the question of ENV function in plants. Demonstration of the biological parameters of ENV-mediated cell-to-cell or plant-to-plant movement could open a range of new applications based on these retroelements. These would be akin to germ-line therapy of mammals using retroviruses in which ENV proteins have been modified to affect host range and targeting (Buchholz et al. 2000). Such strategies would be of particular value in plants, including many crop species, which remain recalcitrant to conventional transformation.
METHODS
PCR and RT–PCR
Template DNA was isolated from leaves as done previously (Vicient et al. 1999). RNA for RT–PCR was isolated with the RNAqueous kit (Ambion 9690) and treated with RNase-free DNase I (Boehringer). The primers for amplification of the env domain consisted of: 5′-CCAAGGTCTATGGGACTTGG AACC-3′ (forward) and 5′-CAAGGGGATTGCCCATACC AATGC-3′ (reverse). Reaction mixtures contained 10 ng of template DNA, 50 pmole each primer, 2.5 mM MgCl2, 1 × buffer (Promega, M190G), 0.2 mM dNTPs, and 0.3 U Taq polymerase (Promega, M186E) in a volume of 25 μL. The PCR reaction program consisted of 5 min at 95°C followed by 31 cycles of 30 sec at 94°C, 2 min at 55°C, 1 min at 72°C, then a final extension for 10 min at 72°C. The RT–PCR was conducted on cDNA prepared with the Qiagen OneStep RT–PCR kit according to the manufacturer's instructions using 1 μg total RNA. The PCR reaction mixture was derived from the kit; the template DNA was produced in the reaction itself during the reverse transcription step. The cycle program consisted of 30 cycles of 45 sec at 94°C, 45 sec at 50°C, and 1 min at 72°C; 10 min at 72°C. Controls for DNA contamination consisted of reactions lacking dNTPs in the reverse transcription step with the nucleotides being added at the beginning of the PCR step.
The PCR and RT–PCR for the rt domain used the following degenerate primers (IUPAC ambiguity codes, I represents inosine): 5′-AARGAYCAYTWYCCIYTICCITT-3′ (forward, p1); 5′-ACCATRAARTGRCAYTTYTCCCARTT-3′ (reverse, p2). The plant accessions and template DNAs were the same as used previously (Suoniemi et al. 1998). The reaction mixtures were as above for the env domain except that they contained 100 pmole primers and 1 U Taq polymerase in a volume of 50 μL. The PCR reaction program consisted of: 5 min at 95°C; 7 cycles of 30 sec at 94°C, 30 sec at 47°C with a decrease of 1°C per cycle, and 3 min at 72°C; 32 cycles of 30 sec at 94°C, 30 sec at 56°C, a warming slope of 16°C in 3 min, and 1 min at 72°C; a final extension for 10 min at 72°C. RT–PCR was conducted as for the env domain with RNA prepared in the same way, using similar controls.
The RT–PCR to detect the spliced RNA used primer p6, an inverse primer in the env domain, 5′-GTTCCTTCCCCTTGG GATCATAGTC-3′, and a direct primer in the Bagy-2 LTR, p5, 5′-TTCGACACTCTTACTTATCGAAAGG-3′. Reaction mixtures were as above for the RT–PCRs for the rt domain. The PCR reaction program was according to the manufacturer for the QIAGEN One Step RT–PCR kit, using an annealing temperature of 50°C, and extension time of 90 sec for 40 cycles.
Cloning and Sequence Analyses
The PCR products were cloned and sequenced as described previously (Vicient et al. 1999) and analyzed by agarose gel electrophoresis under standard conditions (Ausubel et al. 2000). The barley var. Morex BAC clones were from the same set used earlier (Vicient et al. 1999) and were handled as described therein. Sequences were assembled and analyzed using CAP, and alignments were made with theCLUSTALW package, both athttp://www.infobiogen.fr/services/menuserv.html. Sequence searches for additional env-class members were performed with the AdvancedBLAST program using a cutoff value of 0.0001. For those general database entries having a putative translation, we queried the dbEST databases using the TBLASTN program applying a cutoff value of 1.0. All searches were done using the on-line service of the NCBI (http://www.ncbi.nlm.nih.gov/blast/blast.cgi). Relationships between the sequences were analyzed with the distance-based neighbor-joining method available in the TREECON program (Van de Peer and De Wachter 1994).
ENV Protein Motif Analyses
The presence of transmembrane domains was predicted on-line using SOSUI (Hirokawa et al. 1998) athttp://sosui.proteome.bio.tuat.ac.jp/sosuiframe0.html. Their probabilities of occurrence were predicted and graphed usingTMHMM vers. 1.0 throughhttp://genome.cbs.dtu.dk/services/TMHMM/. Hydrophilicity indices were calculated with the Protscale tool using a window of 21 residues by the method of Kyte and Doolittle (1982) available in Expasy(http://www.expasy.ch/cgi-bin/protscale.pl).
IRAP Marker Analyses
The IRAP method has been described previously (Kalendar et al. 1999). Primers matching both ends of the Bagy-2 LTR were used in order to visualize Bagy-2 elements inserted in all three possible orientations with respect to each other. These primers were: 5′-CATGAAAGCATGATGATGCAAAATGG-3′ (forward, E0520), 8 bp from the right end of the LTR, and 5′-TCGAAAGGTCTATGATTGATCCC-3′ (reverse, E0521), 11 bp from the left end of the LTR. Reactions were performed in 20 μL mixtures containing 20 ng of template DNA, 75 mM Tris-HCl (pH 8.8), 20 mM (NH4)2SO4, 1.5 mM MgCl2, 0.01% Tween-20, 200 nM Bagy-2 LTR primers, 0.2 mM dNTPs, and 1.2 U Taq DNA Polymerase. The PCR reaction program consisted of 94°C for 2 min followed by 32 cycles of 94°C for 20 sec, 60°C for 20 sec and 72°C for 2 min, and then a final elongation step of 72°C for 10 min, and was performed in thermocycler (Mastercycler gradient, Eppendorf). To resolve the marker bands, one-fifth of the reaction was analyzed by electrophoresis in 2% agarose (RESolute LE agarose, BIOzym, Landgraaf, The Netherlands) at 80V for 7 h, followed by ethidium bromide staining under standard conditions (Ausubel et al. 2000).
Acknowledgments
We thank Anne-Mari Narvanto for her excellent technical assistance and Joanne Russell (Mylnefield Research Services, SCRI, Invergowrie, Dundee, UK) for the barley cultivars. C.M.V was supported by Academy of Finland Project 44404, R.K. by the European Union Research Directorate under contract QLK5-1999-01499, and A.H.S. in part by an Academy of Finland Senior Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL alan.schulman{at}helsinki.fi; FAX 358-9-191-58952.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.193301.
-
- Received April 19, 2001.
- Accepted October 10, 2001.
- Cold Spring Harbor Laboratory Press








