
Novel Protein Domains and Repeats in Drosophila melanogaster: Insights into Structure, Function, and Evolution
Abstract
Sequence database searching methods such asBLAST, are invaluable for predicting molecular function on the basis of sequence similarities among single regions of proteins. Searches of whole databases however, are not optimized to detect multiple homologous regions within a single polypeptide. Here we have used the prospero algorithm to perform self-comparisons of all predicted Drosophila melanogaster gene products. Predicted repeats, and their homologs from all species, were analyzed further to detect hitherto unappreciated evolutionary relationships. Results included the identification of novel tandem repeats in the human X-linked retinitis pigmentosa type-2 gene product, repeated segments in cystinosin, associated with a defect in cystine transport, and ‘nested’ homologous domains in dysferlin, whose gene is mutated in limb girdle muscular dystrophy. Novel signaling domain families were found that may regulate the microtubule-based cytoskeleton and ubiquitin-mediated proteolysis, respectively. Two families of glycosyl hydrolases were shown to contain internal repetitions that hint at their evolution via a piecemeal, modular approach. In addition, three examples of fruit fly genes were detected with tandem exons that appear to have arisen via internal duplication. These findings demonstrate how completely sequenced genomes can be exploited to further understand the relationships between molecular structure, function, and evolution.
Domains are regions of a protein that form compact three-dimensional structures, and which often evolve independently of one another (Doolittle 1995; Janin and Chothia 1985). Internal duplication within genes appears to have been a prominent evolutionary mechanism for creating functional innovation (Andrade et al. 2001), giving rise to multiple domains or repeats within the same polypeptide (domains are distinguished from repeats in that a domain occurs singly in at least one protein). The discovery of proteins with internal repeats has often been crucial to the detection of a novel homologous family. For example, the realization that pleckstrin contained a domain duplication provided the necessary impetus for the discovery of the pleckstrin homology (PH) domain family (Haslam et al. 1993). Similarly, the identification of WD40-like repeats in β-subunits of G proteins (Fong et al. 1986) preceded extensive investigations into their structures and functions. Consequently, databases such as Pfam (Bateman et al. 2000) and SMART (Schultz et al. 2000), containing curated collections of known domain and repeat families, have proved to be essential for the annotation of protein sequences. However, such resources have not yet succeeded in annotating the majority of eukaryotic sequence with respect to domains and repeats. Much work remains, therefore, in detecting novel families.
To date, identification of domain and repeat families has depended almost exclusively on sequence comparisons of single proteins with nonredundant databases, or all-against-all comparisons of protein sequence sets. Families that have hitherto escaped detection are expected to represent short and/or weakly conserved sequences with similarity scores lying close to background. The background level of similarity encountered in a search is governed by the most statistically significant similarity expected by chance, and depends principally on the number of sequence comparisons made. An all-against-all comparison of N proteins will have a background level P-value N(N−1)/2 times smaller than that observed in a single comparison (Spang and Vingron 2001). Consequently, with the current and rapidly increasing sizes of databases, this strategy may no longer be optimal.
In contrast, we sought to use a search protocol that takes advantage of the observation that genes have frequently evolved by internal duplication. Searching proteins for internal repeats, we argue, is efficient in locating evolutionarily conserved regions as the data set is enriched whereas the number of comparisons made is a factor(N−1)/2 less than before. The background threshold is correspondingly lower so one may detect genuine similarities that were too weak to be significant previously.
The prospero program (Mott 2000), based on earlier theoretical ideas (Karlin and Altschul 1990; Mott and Tribe 1999), is ideal for processing large-scale self-comparisons of protein sequences.Prospero uses a formula that accurately assesses the significance of protein repeat similarities, allowing for the existence of gaps, and furthermore takes into account sequence length and amino acid composition. To a good approximation (Karlin and Altschul 1990;Mott and Tribe 1999), the similarity score S, between unrelated sequences of lengths m, n follows an extreme-value distribution (EVD) depending on two parameters Κ, λ that are functions of the scoring scheme, amino acid frequencies and, to a lesser extent, the sequence lengths: P (S>t) ≈ Κmn exp(−λ t) as t →∞. The distribution of off-diagonal self-comparison scores also approximates to an EVD.Prospero automatically adjusts the values of Κ, λ for each comparison in the calculation of probabilities P. This removes the need to fit the results of simulations to an EVD that would have been time consuming.Our assessment of statistical significance for self-comparisons is slightly conservative. This is because sequence self-comparison is symmetric, such that the expected P-value of the most significant random similarity in Nself-comparisons is about twice that observed in N comparisons between different sequences. Previous analyses have examined general properties of repeats within different genomes (Marcotte et al. 1999; Lavorgna et al. 2001). Here we show thatprospero may be used to gain insights into function and evolution that are not always discernible from standard database searches.
For this study the complete set of predicted proteins (the ‘proteome’) of the fruit fly Drosophila melanogaster (Adams et al. 2000) was targeted for analysis. The genomes of model organisms, including Drosophila, promise valuable insights into biology that extend far beyond narrow phyletic bounds. Genes with a common ancestor (homologs) that occur in two distantly related species, such as an invertebrate and a mammal, are likely to share key roles in physiology and development. This has been borne out by many observations, such as the finding that the majority of human disease genes have orthologous counterparts in the fruit fly (Rubin et al. 2000; Reiter et al. 2001) (orthologs are homologs that have arisen by speciation, rather than by intragenome duplication). In contrast, mutations in genes that are specific to a genus or closely related species appear more likely to present no observable phenotype, at least with currently applied assays (e.g., Gönzcy et al. 2000). Consequently, identifying homologs of human genes encoded within divergent animal genomes, and predicting their functions, is crucial to understanding their roles in human disease and normal cellular processes.
RESULTS
Our analysis of repeats in the 14,226 predicted Drosophilaprotein sequences has contributed to our understanding of the molecular basis of disease and infection, and identified duplications of genes, domains, and exons. The study also demonstrated thatprospero is an appropriate algorithm for detecting homologous protein repeats. Our findings have been set out according to the insights gained into three aspects of biology. First, we describe how repeats in fly proteins led to the identification of novel domain families relevant to human disease and bacterial infection. Second, we discuss the discovery of two novel signaling domain families. Third, we provide evidence for intragene duplication that has given rise to homologous exons and tandem protein repeats. We shall precede these discussions, however, by a description of the general findings of the study.
General Trends
Of 14,226 fruit fly sequences, prospero identified 1656 that contained (at least) a pair of internal repeats according to the criteria used for their detection (Fig. 1); of these 523 represented novel repeats (see Methods). Just over half (591) of these 523 × 2 = 1046 repeat sequences could be grouped, using single-linkage clustering of BLAST alignments and a minimum threshold of 50 bits, into clusters containing at least two members (see Methods for details), leaving the remaining 455 sequences as members of the ‘orphan’ set. All groups and all orphan sequences were analyzed in-depth using iterative sequence database search methods. This led to approximately one-fifth of groups and orphan sequences being identified as containing repeated compositionally biased repeats, including transmembrane sequences, that escaped masking by the SEG algorithm (Wootton and Federhen 1993). In addition, 23 groups (10%) and 90 orphan sequences (20%) were found to contain short repeats of <30 amino acids that are unlikely to form globular domain structures. The remaining 161 groups and 287 orphan sequences represented longer repeats, likely to form compact domains. Approximately half of these, however, were repeats that were detected in fewer than four fly proteins. The small fly-specific families, as well as those representing short repeats and compositionally biased repeats, were not considered further.
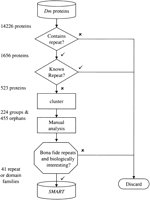
Flow diagram of the Drosophila repeat discovery protocol. Sixteen hundred and fifty-six of 14,226 predicted Drosophilaproteins were found to contain repeats (see Methods), of which 523 represented previously unknown repeats. Following a clustering step, these were partitioned into 224 groups and 455 orphans that were subjected individually to manual analyses. This resulted in the identification of 41 families of repeats and domains whose multiple alignments have been deposited in the SMART database (http://www.smart.embl-heidelberg.de).
Exhaustive analysis of the remaining 94 groups and 142 orphan sequences identified 41 families (Table 1) of which, 27 were unrecognized previously, or greatly expanded, families of repeats or domains, and 14 represented additions to previously known families. Eight of the previously unrecognized families contained no members from organisms other than Drosophila. All the families' alignments were constructed and submitted to the SMART database. Internally repetitive proteins were not found to be specific to particular cellular roles or particular domain types. For example, tandem repeats were found in inorganic phosphate, dicarboxylate, sugar, and amino acid transporters (fly proteins CG11682, CG4961, CG6645, and CG11806, respectively), and enzymes (below).
Domain and Repeat Families Identified
Repeats in Enzymes
More than a dozen different domain families of enzymes were found repeated in fly proteins. Thirteen fly proteins, for example, were found with two, and one (CG8215) with four, tandem repeats homologous to trypsin-like serine proteases. Of these 14, all but two conserved the His/Asp/Ser catalytic triad. The exceptions (CG9898 and CG8555) are likely to lack proteolytic activity. Repeated trypsin-like domains are unusual, being entirely absent in known mammalian proteins, and found only once in Xenopus (Lindsay et al. 1999) and once inSaccharomyces cerevisiae (Ponting 1997).
In a few instances, further analysis of repeat-containing proteins resulted in predictions of enzymatic activity relevant to pharmacology. For example, CG10183 contains two Cys-rich domains that are otherwise present as single copies in members of a family of transmembrane proteins. This family includes the products of genes nrf-6 andndg-4 that are mutated in Caenorhabditis elegansresistant to fluoxetine (prozac), via a mechanism that is distinct from the inhibition of serotonin reuptake (Choy and Thomas 1999). Analysis of this family demonstrated that these proteins are homologs oftrans-acylation enzymes. Indeed, worm genes (C06B3.2 and F09B9.1) described as homologs of nrf-6 and ndg-4 in the primary literature (Choy and Thomas 1999) have been described previously as trans-acylases (Slauch et al. 1996). Thus, these findings provide the first suggestion that trans-acylation is involved in the nonserotonergic effect of fluoxetine in C. elegans.
Repeats in Human Disease-Associated Proteins
Tandem Repeats in Human X-Linked Retinitis Pigmentosa Type 2 (XRP2)
This study found four novel domain types that provide insights into human disease. The first of such cases concerns tandem repeats in the product of the human X-linked retinitis pigmentosa type-2 (XRP2) gene. This is mutated in patients with a severe form of retinal degeneration that causes night blindness and constricted visual fields (Schwahn et al. 1998). These repeats could not be detected by querying databases with the complete sequences and PSI-BLAST.
The fly CAPT (an adenylyl cyclase-associated protein [CAP] homolog; GeneInfo code 7296114) was predicted to contain tandem repeats with aprospero P-value of 5.4 × 10−6. Although CAPs and XRP2 were not previously thought to be homologs, this is clearly the case. A PSI-BLAST search of current databases using human XRP2 detected Arabidopsis thaliana CAP (the T4L20.70 gene product) in the second iteration with anE-value of 9 × 10−4. These homologous regions encompass the repeats detected in the fly CAPT protein, and two missense mutations in XRP2 that were found in patients with this disease (Fig. 2) (Schwahn et al. 1998;Mears et al. 1999).
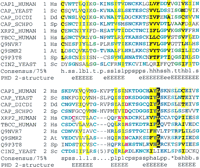
Multiple sequence alignment of tandem duplication of CAP-related protein (CARP) domains in adenylyl cyclase-associated proteins (CAPs) and other proteins presented using CHROMA(Goodstadt and Ponting 2001). The tandem contiguous CARP domain repeats are shown in two successive tiers in the following order (amino acid numbers and GeneInfo codes): Homo sapiens (Hs) CAP1 (356–431: 399184); Saccharomyces cerevisiae (Sc) CAP (Srv2p) (406–481: 134897); Dictyostelium discoideum (Dd) CAP (344–419: 1705592); Schizosaccharomyces pombe (Sp) CAP (432–507: 543928); H. sapiens Xrp2 (67–140: 6831708); H. TCC (211–284; 6831693); H. sapiens FLJ10560 (337–410: 8922517);Arabidopsis thaliana (At) T5J17.90 (206–279: 7487709),Schizosaccharomyces pombe SPAC328.08c (151–223: 8894859); andS. cerevisiae Cin2p (130–208: 1168956). Underlined residues in human RP2 are mutated in individuals with X-linked retinitis pigmentosa (Schwahn et al. 1998; Mears et al. 1999). Secondary structures predicted (Rost and Sander 1993) at expected accuracies of >82% (E) or >72% (e) are indicated below the alignment (E/e, extended or β-strand structure).
XRP2 was previously predicted to have a role in the tubulin folding pathway because of its significant sequence similarity to tubulin-specific chaperone cofactor C (TCC) in the tandem repeat region (Schwahn et al. 1998). Yeast Cin2p is also predicted to contain CAP-, XRP2- and TCC-like repeats on the basis of a PSI-BLASTsearch initiated using human TCC that identified Cin2p (chromosome instability 2 protein) after two rounds withE = 10−2, and knowing that TBCC and Cin2p have comparable functions in mammals and yeast, respectively. Previous studies have identified yeast homologs of all mammalian tubulin-specific chaperone cofactors except for TCC (Tian et al. 1996); mammalian homologs of all yeast CIN genes, except CIN2 have also been proposed (Fleming et al. 2000).
The homologous repeats in CAPs, TCC, and Cin2p suggest a previously unforeseen role for CAPs in tubulin biogenesis. Until now, the roles of the multifunctional CAPs had been ascribed to the biogenesis of the actin-based cytoskeleton (Zelicof et al. 1996), rather than the correct folding of tubulins, the major constituents of microtubules. However, it should be noted that yeast strains with disruptions in the CAP gene show aberrant staining of tubulin as well as actin, and this phenotype has been mapped to the repeat-containing C-terminal region of CAP (Gerst et al. 1991).
Tandem Repeats in Muscular Dystrophy-Linked Genes
Muscular dystrophy is a heterogeneous group of mammalian hereditary diseases that cause progressive muscle weakness. Of these diseases, the most common is X-linked Duchenne muscular dystrophy caused by mutations in the dystrophin gene, DMD. Loss of the dystrophin-associated protein dystroglycan in mice causes a loss of muscle function similar to that in human muscular dystrophy (Cote et al. 1999). α-Dystroglycan (αDG) and β-dystroglycan (βDG) are products of the same dystroglycan (dystrophin-associated glycoprotein-1) gene, generated posttranslationally via proteolytic cleavage. Two tandemly repeated regions (amino acids 649–832 and 880–1061) ofDrosophila CG18250, the presumed fly ortholog of human DG, were identified by prospero as homologous (P = 2.5 × 10−30). In BLASTsearches, these regions both show significant similarity (E = 5 × 10−10 and 2 × 10−16) to a single region of human DG (approximately amino acids 494–674) that spans the αDG/βDG boundary. This implies differences in the molecular functions of the presumed fly and human DG orthologs as proteolysis of fly CG18250 in the two positions corresponding to the αDG/βDG boundary in mammals would result in two, rather than one, soluble extracellular proteins in addition to a single membrane-associated βDG-like protein.
The dysferlin gene is mutated in limb girdle muscular dystrophy (Bashir et al. 1998; Liu et al. 1998). Our search protocol revealed a pair of repeats (prospero P = 2.52 × 10−10) in a fly protein, CG6468 that, on further analysis, could be decomposed into five repeating units homologous to the β-propeller blades of Physarum polycephalumtectonins (Huh et al. 1998). Analysis of the remainder of the CG6468 sequence then revealed a ‘DysF’ domain of unknown function that is homologous to two domains in dysferlin (Fig.3). Interestingly, these two copies are not arranged in tandem, as is most usual with repeats. Rather, one DysF domain is inserted within a second DysF domain. This evolutionary scenario is extremely rare, happening to our knowledge only with PH domains in myosin X (Berg et al. 2000). The nesting of homologous domains one-within-another might be considered, for example, to reflect exacting constraints on binding sites, or else might represent a serendipitous event.
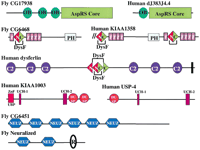
Schematic representation, shown approximately to scale, of the domain architectures of some of the proteins containing repeats or domains that are discussed in the text. Abbreviations: AAA, ATPases associated with a variety of cellular activities; AspRS core, the catalytic portion of aspartyl tRNA synthetases; Bbox, B-box type zinc finger; C2, Protein kinase C conserved region 2 (CalB) domain; F, F-box domain; OB, oligonucleotide/oligosaccharide binding fold; PH, pleckstrin homology domain; R, RING finger domain; T, tectonin-like β-propeller repeats; UCH-1, UCH-2, Ubiquitin carboxyl-terminal hydrolases family 2 (two conserved regions); WD, β-propeller repeat with conserved Trp (W) and Asp (D) residues; ZnF_UBP, Ubiquitin carboxy-terminal hydrolase-like zinc finger; ZnF_UBR1, domain that is involved in recognition of N-end rule substrates in yeast Ubr1p. Solid vertical lines represent predicted transmembrane helices. Double forward slash lines indicate that the protein sequence is N-terminally truncated.
Repeats in Cystinosin, the Infantile Nephropathic Cystinosis Gene Product
Infantile nephropathic cystinosis is a lysosomal storage disorder caused by a defect in cystine transport across the lysosomal membrane. Mutations in the CTNS gene have been identified in patients with this disease (Town et al.1998). Cystinosin, the CTNS gene product, is a seven-transmembrane protein whose molecular function is unknown. Our studies show that cystinosin, and many other membrane proteins, possess a pair of repeats each spanning two transmembrane helices connected by a loop (Fig. 4a). After initial submission of this manuscript, others have reached a similar conclusion (Zhai et al. 2001). Drosophila CG17119 (a likely fly ortholog of human cystinosin) was found to possess these repeats with P = 3.1 × 10−5. Within these repeats, a proline and glutamine amino acid pair is prominently conserved (Fig. 4a).



Multiple sequence alignment of (a) transmembrane-containing segments in cystinosin (CTNS) homologs, (b) LITAF (lipopolysaccharide-induced tumor necrosis factor α factor) homologs, and (c) Myb-like repeats in timeless (TIM) homologs aligned against Xenopus laevis (Xl) B-Myb (GeneInfo: 6226654), presented using CHROMA (Goodstadt and Ponting 2001). (a) CTNS-like repeats are shown in pairs in the following order (amino acid numbers and GeneInfo codes): Homo sapiensCTNS (140–171 and 279–310: 4826682); H. sapiens SL15 (56–87 and 167–198: 4759110); Arabidopsis thaliana SL15 (44–75 and 156–187: 8885552); Saccharomyces cerevisiae Endoplasmic reticulum defect suppressor 1 (ERS1p), (18–49 and 168–199: 6319919);Drosophila melanogaster CG17119 (145–176 and 284–315: 7300949); and, D. melanogaster CG3792 (SL15) (51–82 and 162–193: 10728573). Missense mutations in cystinosin (Attard et al. 1999) are shown in red and are underlined. A YFPQA pentapeptide in cystinosin that when mutated resulted in protein targeting abnormalities (Cherqui et al. 2001), has been underlined. (b) Hydrophobic residues that are likely to insert into membranes are relatively ill-conserved and have been replaced in the alignment by their respective numbers. GeneInfo codes and amino acid limits are given following the alignment. (c) The following sequences are shown: D. melanogaster TIM (913–971: 6175063) 913–971;Rattus norvegicus TIM (816–873 and 883–937: 7514104) ;D. melanogaster TIM2 (824–883 and 896–950: 8133124);Caenorhabditis elegans Y75B8A.22 (970–1029, 1038–1099, and 1106–1168: 7510480); and, A. thaliana TIM (847–903 and 928–984: 10177105). The secondary structures known from the B-Myb crystal structure (PDB code 1MSE) are shown beneath the alignment together with the predicted (Rost and Sander 1993) secondary structures of the TIM repeats at expected accuracies of >82% (H) or >72% (h); H/h, helical structure. Species abbreviations used: At, A. thaliana; Ce, C. elegans; Dm, D. melanogaster; Hs, H. sapiens; Mm, Mus musculus; Pf, Plasmodium falciparum; Rn, R. norvegicus; and Sc, S. cerevisiae. (Figure continues on next page.)
The reason for the conservation of the transmembrane helix–loop–helix repeats in general, and this ‘PQ motif’ in particular, was unclear until recent mutational studies by Cherqui et al. (2001). These investigators found that the PQ-containing loop of repeat 2 is critical for the localization of cystinosin to lysosomes. We identified 25 eukaryotic proteins that contain the PQ motif. One of these, SL15, is a suppressor of Chinese hamster ovary cell glycosylation mutations and its protein is localized to the endoplasmic reticulum (Ware and Lehrman 1996). Thus, the PQ motif appears not to be a general lysosome-targeting motif; rather, it is likely to possess a more general function. Most probably this involves a glutamine residue, as this is strongly conserved among 25 eukaryotic proteins (Fig. 4a).
Disease and Infection: Lipopolysaccharide-Associated Domains
Vertebrates and invertebrates have evolved molecular mechanisms to defend against infection by microorganisms. These include the response to lipopolysaccharides (LPS) expressed on the surface of bacteria. One human gene induced by LPS is LPS-induced tumor necrosis factor α factor (LITAF) (Myokai et al. 1999). In our searches for repeats inDrosophila proteins, one (CG13515) had a tandem duplication that was shown, by hidden Markov model (HMM)-based database searches, to be homologous to LITAF. Approximately a dozen of these domains were detected in both C. elegans and Drosophila, and in a single additional human protein (transmembrane protein Il, GenInfo identifier 11493639). The LITAF-like domain is unusual in that it contains an N-terminal CxxC ‘knuckle’ followed by a long (∼25 amino acids) hydrophobic region, and a C-terminal (H)xCxxC knuckle (Fig. 4b). Because both these knuckles are highly characteristic ofintracellular Zn2+-binding domains, and the N-terminal region of one LITAF-like domain-containing protein is thought to bind the intracellular molecule Nedd4 (Jolliffe et al. 2000) it appears clear that the hydrophobic region does not span the membrane. Rather it is likely to be a region that inserts into, but does not traverse, membranes and which brings together the N- and C-terminal CxxC knuckles to form a compact Zn2+-binding structure. Because LITAF (also known as PIG7) is induced by p53 (Polyak et al. 1997), it is possible that these LITAF-like domain proteins are involved in LPS-induced onset of apoptosis.
Repeated copies of a polysaccharide biosynthesis-associated domain were found in fly CG17138. Single copies of this domain have been documented previously in mouse EP58, the Dichelobacter nodosuslipopolysaccharide biosynthesis gene LpsA product, and in theCryptococcus neoformans polysaccharide capsule-associated geneCAP10 product (Kimata et al. 2000). A PSI-BLASTsearch using the VGE server (http://www.vge.ac.uk/blast/psiblast.html) and amino acids 131–502 of EP58 identified a putative homolog of these domains in Bacillus anthracis after two rounds (E = 7 × 10−3). This proposed homolog (GBant64-5) is a member of family 1 nucleotide-diphospho-sugar glycosyltransferases (Campbell et al. 1997). Similarly, the top scoring, nonsignificant, sequence in a HMM-search of databases was also a family 1 glycosyltransferase (Synechocystis sp. slr1063; E = 7.3). This family is characterized by acidic active site residues separated by seven and sometimes eight other amino acids: E-X(7,8)-E. The first of these acidic residues appears to be present, as an aspartic acid, in most of the CG17138/LpsA/CAP10-like domains, whereas the second is absent (data not shown). It is concluded that this family probably represents a highly divergent set of glycosyltransferases. This would be consistent with the proposed polysaccharide processing functions of CAP10 and LpsA. Because several inherited diseases are associated with deficiencies in glycosylation enzymes (for review, seeAebi and Hennet 2001), it is possible that deficiencies of the ER-associated protein EP58 and other human CAP10 homologs might also be linked to human disease.
New Signaling Domain Families
In addition to the discovery of homologous repeats in RP2 and CAPs described above, we have identified two other novel signaling domain families.
Tandem repeats were found in the fly neuralized gene product, involved in development of the central and peripheral nervous system (Boulianne et al. 1991). Several other fly, worm, and mammalian proteins were found to contain between one and six of these repeats, which we refer to as ‘NEUZ’ domains (Fig. 3). The NEUZ domains' functions are not known, although they do partly resemble SPRY domains (Ponting et al. 1997). Querying Pfam with fly CG6451 results in a prediction of an N-terminal SPRY domain (E = 0.55) that entirely encompasses the most N-terminal of six NEUZ domains. This possibility is consistent with the fact that both SPRY and NEUZ domains are found to cooccur in proteins with RING fingers. If NEUZ and SPRY domains are homologous it is possible that NEUZ possess microtubule-binding functions, similar to those proposed for SPRY domains (Cox et al. 2000).
A pair of homologous domains was found at the C terminus of CG8494 and its presumed human ortholog KIAA1003 (Fig. 3), which are ubiquitin-specific proteases (USPs). A similar ‘DUSP’ domain is found at the N terminus of ubiquitin-specific protease-4 (USP-4). By their cooccurrence with USP hydrolase domains, it is likely that DUSP domains regulate USP-mediated proteolysis (Baker et al. 1992).
Protein Evolution by Internal Domain Duplication: Timeless and Aspartate-tRNA Ligase
We identified a pair of tandem repeats in the fly TIM-2gene product (also known as Timeless-2, Timeout, or CG8148) (prospero P = 9.1 × 10−6). The fly TIM-2 repeats also cannot be identified usingPSI-BLAST. Further database searches usingHMMER2 identified homologs of the fly TIM-2 tandem repeat once in fly TIM (the timeless gene product), twice in rat TIM, and three times in C. elegans Y75B8A.22 (Fig. 4c). C. elegansY75B8A.22 appears to be the ortholog of fly TIM-2, yet it contains one extra tandem repeat. Secondary structure predictions indicate that these repeats adopt a three-α-helix fold. This is highly reminiscent of the Myb fold (Ogata et al. 1992) (Fig. 4c). Indeed, with a search of databases using a HMM derived from the alignment of TIM repeats (Fig.4c), Xenopus laevis B-Myb was the second highest scoring sequence, albeit with a nonsignificant E-value (E = 9.4).
Fly TIM is essential for circadian clock function. At dusk it associates with Per, the product of the period gene, and increasingly as night falls translocates to the nucleus, where the heterodimer represses transcription of TIM and periodgenes. Association with Per occurs in a region of TIM (van der Horst et al. 1999) that is N-terminal to its Myb-like repeat. Three-α-helix domains, and Myb domains in particular, might be thought to possess DNA-binding properties. TIM, however, is not known to possess affinity for DNA, but it does inhibit the DNA-binding activities of other proteins with the same domain architectures as Per (Lee et al. 1999). This suggests that the TIM repeats might be involved in binding Per-like proteins. This is not incompatible with the prediction that TIM repeats might be Myb domain homologs, as some Myb domains are known to bind protein (Cutler et al. 1998).
We also observed different domain numbers between aspartyl t-RNA synthetases (AspRS) orthologs. Drosophila AspRS (CG17938) contains three repeats of an OB-fold tRNA anticodon-recognition domain, whereas most other eukaryotic AspRS enzymes, such as that of yeast (Ruff et al. 1991), contain only one (Fig. 3). In this case, it remains to be determined whether the fly AspRS obtains additional advantage in possessing three consecutive versions of a domain that exists in only one copy in orthologous AspRSs.
Rapid Evolution of Domain Families within Limited Taxonomic Ranges
This study frequently identified arthropod-specific expansions of domain families. These included Adf1- and Myb-like DNA-binding domains (Cutler et al. 1998) in 55 fly proteins, juvenile hormone-binding protein domains (Lerro and Prestwich 1990) in 33 proteins, and pheromone-binding protein-like domains (Raming et al. 1989) in 28 proteins. The large expansions of these domain families in theDrosophila proteome suggest key roles in fly physiology and evolution.
The CLIP Domain
The CLIP domain is perhaps the most intriguing of those found in narrow phyletic ranges. First identified in the horseshoe crab,Tachypleus tridentus, the domain is found in varying copy numbers (from one to five in Drosophila proteins), but always N-terminal to trypsin-like serine protease domains (for review, seeJiang and Kanost 2000). The region is characterized by six conserved cysteines. The species distribution of known representatives, including crustaceans, the horseshoe crab, and a variety of insects, suggests that the domain is specific to arthropods. Using a combination of HMM searches and pattern analysis of regions N-terminal to trypsin-like serine protease domains we identified potential CLIP domains in ⋝36Drosophila proteins. Precise counts are difficult, as there is little sequence conservation beyond the six cysteines, and the spacing of cysteines is highly variable. CLIP domain-containing serine proteases are known to play important roles in Drosophiladevelopment, including the Snake and Easter proteins, which form part of a cascade initiating dorsal-ventral patterning. The CLIP domain is also found in prophenoloxidase activating enzymes involved in the innate immune response. The prophenoloxidase activating enzyme from the freshwater crayfish, Pacifastacus leniusculus contains an N-terminal CLIP domain that has been shown to have antimicrobial activity (Wang et al. 2001), and CLIP-containing serine proteases from other arthropods have also been implicated in other innate immune responses. Factor B of the Tachypleus blood coagulation cascade is a similar CLIP domain-containing serine protease, and this cascade is linked to prophenol oxidase activation (Nagai and Kawabata 2000). It seems likely that the CLIP domain is responsible for mediating specific protein–protein interactions, and as such is useful for regulating cascades of serine protease activities (Jiang and Kanost 2000) similar to those of the vertebrate blood coagulation system (Iwanaga et al. 1992).
Although current data suggest the CLIP domain is specific to arthropods, this could simply be due to rapid evolution resulting in detectable sequence similarity to proteins in other metazoan lineages being lost. An interesting parallel case to this is the structural similarity apparent between coagulogen, the clotting protein from horseshoe crab, and vertebrate nerve growth factor-like (NGF) proteins (Bergner et al. 1996). Here it is the NGF domain that appears, at the sequence level, to be specific to vertebrates, although comparison of structures suggests that coagulogen is indeed homologous. Thus, it seems plausible that the CLIP domain is homologous to one of several families of cysteine-rich domains found N-terminal to vertebrate serine proteases.
Evolution of Domains by Internal Duplication: Glycosyl Hydrolases
Recent results have shown that several symmetrical structures previously thought of as distinct globular domains contain patterns of sequence conservation that suggest they evolved from smaller repeating units (Coles et al. 1999; Lang et al. 2000; Ponting and Russell 2000). As well as being relevant to understanding the evolution of these protein fold families, such results are important because they raise the possibility that the repeating sequence segment may later be found singly, perhaps embedded within different folds. This study identified repeats in αα toroids and β-helix-containing proteins.
Repeats in αα Toroids
The product of the Drosophila gene CG18799 was found to contain an internal repeat (prospero P-value of 7 × 10−6). The protein is highly homologous to a class I α1,2-mannosidase of known structure from S. cerevisiae(Vallée et al. 2000). This structure has an (αα)7barrel topology, and is classified in Scop (Lo Conte et al. 2000) as a seven hairpin glycosyltransferase of the αα toroid fold class. Inspection of the alignment of CG18799 to the yeast protein sequence showed that the repeats found in the Drosophila protein map to the third and fourth αα hairpins (Fig.5a). Some, but not all, of the αα hairpins are interrupted by a pair of antiparallel β-strands. Thus it appears likely that both class I α1,2-mannosidases evolved from multiple internal repetition of an ancestral αα or αββα structure, and also that sequence evidence for the common ancestry of the internal repeats is now only evident in the third and fourth αα hairpins.
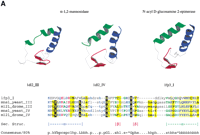

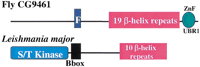
(a) αα repeats found in αα toroids. The third and fourth repeats in yeast α-1,2-mannosidase (PDB code: 1DL2) are detectable by sequence in the equivalent Drosophila protein. The repeating unit is also structurally equivalent to that found in the αα6 class of proteins, such as N-acyl D-glucosamine 2-epimerase (PDB code 1FP3), indicating the two classes of proteins shared an ancestor (see text for details). The sequence alignment produced by aligning the structures of the individual repeats is shown beneath the alignment. Structurally equivalent residues, as defined by the STAMP program (Russell and Barton 1992) are upper case. Nonequivalent regions are lower case. (b) The 3D structure of Aspergillus niger endopolygalacturonase II (PDB code 1CZF). For clarity, the three β-strands in each repeat are colored blue, red, and green. Continuous strands have been split in two at a region corresponding to a pronounced bend in the polypeptide chain, as defined by the DSSP program. Also shown is a multiple sequence alignment of β-helix repeats in A. nigerendopolygalacturonase II (GeneInfo number 6435555) and Drosophila melanogaster CG9461 (GeneInfo number 7299263) represented usingCHROMA (Goodstadt and Ponting 2001). Colored arrows under the alignment correspond to the β-strands in the 3D structure. Asn residues that might form an Asn ladder (see text) are shown as white-on-black. GeneInfo codes for the repeats in PDB:1CZFA andD. melanogaster CG9461 are 6435555 and 7299263; the repeats represent 1CZFA 156–184, 187–206, 209–227, 238–257, 267–287 and 301–327; and CG9461 624–644, 650–670, 673–693, 696–716, 719–739, 742–762, 765–785, 788–808, 811–831, 834–854, 857–877, 880–900, 903–923, 926–946, 949–969, 972–992, 995–1015, 1018–1038, 1041–1061, and 1064–1083.
A similar repeating unit is found in another class of glycosyltransferase that adopts an (αα)6 barrel topology (e.g., the structure of N-acyl-D-glucosamine 2-epimerase, Itoh et al. 2000). Both (αα)6 and (αα)7 barrels are members of the generic α/α toroid fold class of Scop. However, unlike other members of this fold family, the two types of barrel both share the characteristic extended linker region between the pairs of helices that form the αα units (see Fig. 5a). This structural detail, taken together with related functions, suggests that the repeating units composing the two barrel classes share a common ancestor. Whether the (αα)7 class of barrel is a recent derivative of the (αα)6 class, or whether both represent ancient forms derived from the same ancestral peptide repeat is not easily resolved. Structural alignment does not provide overwhelming evidence that any of the individual (αα) units found in α1,2-mannosidase is a recent insertion in the (αα)6 fold.
Repeats in Parallel β-Helices
A different glycosyl hydrolase provided a further example of repeats that assemble into a single protein domain. Following the identification of repeats in CG9461, iterative sequence database searches using HMMER and an E-value inclusion threshold of 0.1, predicted sequence-similar repeats (Fig. 5b) in mannuronan C-5-epimerases and nosD, a periplasmic protein that may facilitate insertion of copper into a nitrous oxide reductase (Holloway et al. 1996). These searches also revealed that such repeats are homologous to those found in parallel β-helices in pectate lyases,Aspergillus aculeatus rhamnogalacturonase A, and P22-phage tailspike protein (for review, see Jenkins et al. 1998).
Each turn in the β-helix corresponds to a single sequence repeat that contains three or four β-strands. In pectate lyases, the conservation of asparagines in sequential repeats results in the stacking of their side chains in an ‘asparagine ladder.’ Conservation of asparagine in the CG9461 repeats indicates that this side-chain stacking also occurs in its β-helix (Fig. 5b). In bacterial mannuronan C-5-epimerase, the parallel β-helix predicted here corresponds to the so-called ‘A domain’ that possesses epimerase and Ca2+-binding activities in isolation (Ertesvåg and Valla 1999). This compares well with the enzymatic activities of most other β-helix proteins, and the Ca2+-binding function of some pectate lyases.
Many of these β-helix fold proteins are enzymes that bind α-galactose-containing polymers. Thus we predict thatDrosophila CG9461, and its animal orthologs, might be polysaccharide-binding proteins or even glycosyl hydrolases. This would be unusual because its domain architecture (Fig. 3) implicates these molecules in the intracellular proteolytic pathway of the ubiquitin system, rather than in the modification of polysaccharides. Nevertheless, the animal CG9461 orthologs represent, to our knowledge, the first examples of a parallel β-helix protein described in metazoa.
Evolution of Gene Multifunctionality by Exon Duplication
On three separate occasions we detected tandem repeats in predicted gene products that did not correspond to either domains or structural repeats. Rather they represented degenerate repeats encoded by tandem exons within the same gene. The Genewise program (Birney,http://www.sanger.ac.uk/Wise2) was used to align the protein sequences to genomic DNA. This demonstrated in each of the three cases that the protein repeat was apparent as repeats in the genomic DNA, and each repeated segment corresponded to an individual exon (Fig.6). In each case, the sequences of the pairs of exons were nonidentical, making it unlikely that the genomic DNA had been duplicated artefactually in the assembly procedure. Multiple alignment and phylogenetic analysis suggested that in all cases the repeated exons arose via duplication of an exon within the gene, rather than a later independent insertion into the protein (data not shown).
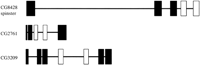
Schematic representation of the gene structures of CG8428, CG2761, and CG3209. These were calculated by aligning the predicted protein sequences to the corresponding genomic DNA using theGeneWise algorithm of Birney (http://www.sanger.ac.uk/Software/Wise2). Genes are shown to scale in the 5′–3′ direction. Exons are represented as vertical boxes, introns by horizontal lines. Homologous exon pairs are shown in white, other exons in black.
In all three genes, the repeated exon appears to have functional significance. CG8428 encodes a hypothetical protein, predicted to be a sugar transporter (Pfam family PF00083), where each repeated exon corresponds to three transmembrane helices. The CG8428 sequence essentially encompasses the products of each of the multiple distinct variants of the spinster gene but, in each case, the sequences of the spinster transcripts include only one of the two repeated exons of CG8428. CG2761 encodes a NodB-like polysaccharide deacetylase (Pfam family PF01522). The repeated exons each include at least one residue (D101 and D151) that is likely, based on amino acid conservation, to be within the active site. The repeat in CG3209 is part of the N-terminal portion of a phosphate acyltransferase domain (Pfam family PF01553), again including residues (H256 and D261; H334 and D339) predicted to be in the active site (Neuwald 1997).
In none of these three cases do the tandem repeats correspond to structural domains. Consequently, and in contrast to their database annotations, it seems unlikely that any of these exon pairs occur as a pair in the same transcript. Instead, we suggest that each exon pair represents ‘either/or’ choices for the generation of alternatively spliced products, each with differing substrate specificities. Indeed, following the original submission of this manuscript Nakano et al. (2001) reported that multiple spinster (CG8428) transcripts are generated by such an either/or choice mechanism. As more cDNA sequences become available, and a better picture of theDrosophila transcriptome is presented, the bases for the multifunctionality of alternatively spliced genes should become clearer.
These cases are similar to the either/or splicing seen in theDrosophila homolog of Down syndrome cell adhesion molecule (DSCAM). For this gene, four out of 24 exons are variable (but within each of the four exon groups, alternative sequences are homologous) potentially leading to >38,000 splice forms (Schmucker et al. 2000). Similar processing has also been postulated as a possible explanation for the genomic organization of neural cadherin-like cell adhesion genes in humans (Wu and Maniatis 1999).
DISCUSSION
The results of this study demonstrate that repeat detection is a viable and fruitful approach in the discovery of novel families. Whereas whole database searches, as performed by the popularBLAST suite of programs, are often successful in predicting domain and repeat families, many of the findings in this study were not detectable using this approach. The method employed here detected many different types of repeats including some 30%–40% of repeats that represent either compositionally biased sequences or else nonglobular regions. Exclusion of these biologically less informative repeats necessitated manual curation of the set.
Three findings that were identified in this study were not discussed at length, due to their independent discovery by others elsewhere. The first of these was the finding that E-Z repeats (Dolganov and Grossman 1999) are outlier members of the HEAT repeat family (Neuwald and Hirano 2000). Secondly, a repeated domain in fly Deltex, a cytoplasmic modulator of Notch-signaling in animals (Xu and Artavanis-Tsakonas 1990; Matsuno et al. 1998) was also found in proteins involved in the ubiquitin-mediated proteolysis pathway and ADP ribosylation (Aravind 2001). Lastly, the family of lysyl/prolyl hydroxylases was found to be more extensive than realized previously (Aravind and Koonin 2001). The latter finding is poised to attain greater significance following the recent identification of proline hydroxylation as the O2-dependent posttranslational modification mechanism that targets some proteins for degradation (Ivan et al. 2001; Jaakkola et al. 2001).
Of the repeats found in Drosophila as well as one other complete genome, three (MADF, NRF, and DM13) are found inDrosophila and C. elegans but not human, and one (THEG) is found in Drosophila and human, but not C. elegans. In the absence of agreement on the possibility of anecdysozoa clade (which would unite Drosophila andC. elegans in a clade, with human as an outgroup) (Aguinaldo et al. 1997; International Human Genome Sequencing Consortium 2001), this study was unable to distinguish between the distinct evolutionary scenarios of loss in one lineage, versus gain in the other lineages.
Findings from this study highlighted the susceptibility of proteins throughout evolution to multiple distinct types of duplication: duplication of exons, such as those in spinster; duplication of domains, such as the tandem repeats in Timeless and aspartate-tRNA ligase; and duplication of whole proteins, for example the fly trypsin homologs, one of numerous families that have been subjected to lineage-specific expansion. A fourth duplication type results in short tandemly repeated sequences that assemble into larger and compact protein domains. The discovery of repeats in αα toroids, for example, implies that these enzymes arose from ancestors that contained fewer such repeats, and even earlier ancestors that contained only a single such ‘repeat.’ This indicates that this common ancestor associated into an oligomer to form a functional and structural stable unit. Although the duplication of domains is obviously a major factor shaping the architectures of metazoan proteins, the existence of tandemly repeated homologous exons, and multiple repeat-containing domains, emphasizes that smaller ‘sub-domain’ units have also provided the raw material used in gene evolution.
With regard to either/or splicing, it is relevant to ask how common this phenomenon is likely to be. Although this study only detected three examples of exon duplication, there are several reasons why the phenomenon might be more widespread. Firstly, in our analysis we discarded sequence homologs that are Drosophila-specific. Exon duplication occurring in such sequences would obviously not be detected under our protocol. Secondly, if the repeated exon is found in a protein with repeated well-described domains, it will be screened out in our procedure (as our primary interest in this study has been the identification of new domains). Finally, we analyzed a predicted protein set. These sets are created using the evidence of ESTs and full-length cDNAs to predict splicing patterns of genes. Thus, if two exons of the same gene are mutually exclusive, they may not appear in the same transcript (i.e., predicted protein) and again, will not be detected by our method. Rather than being a problem, this will be the situation for correctly predicted transcripts. For analyses such as these, it could be countered by producing ‘complete transcripts’ in silico, where a notional peptide would be constructed from all the exons of a gene (taking no account of splicing patterns), and searching for repeats in this sequence set.
An unresolved question is the extent to which similar genetic mechanisms could be responsible for all four of the types of repetition identified above. Repeats could arise via multiple independent insertions of a DNA encoding a specific domain into a particular gene. Alternatively, they could arise from slippage occurring during DNA replication. Theoretically these two scenarios are distinguishable by phylogenetic analysis. In practice however, such analyses suffer from a lack of resolution resulting from the short lengths and significant sequence divergence among repeats.
This study of Drosophila proteins has implications that extend far beyond an understanding of fruit fly biology. The detection of these repeats has led directly to valuable insights into the molecular basis of human disease, and other general aspects of protein evolution and function. Analyses of repeats in other completely sequenced genomes are likely to lead to similar advances.
METHODS
Automated Analysis
The complete set of N = 14,226 proteins originally predicted from the D. melanogaster genome (Adams et al. 2000) was masked for low complexity and coiled-coil regions using default settings (Lupas et al. 1991; Wootton and Federhen 1993). Masking was employed to reduce the number of repeats found that simply correspond to compositionally biased regions including transmembrane helices and coiled coils. Internal sequence repeats were predicted by self-comparison of each Drosophila sequence, using theprospero program of Mott (2000). Rather than examining all repeats detected, only the highest scoring suboptimal repeat was considered. Matches were regarded as significant if their estimated probabilities of occurring by chance P were <10−4(see Mott 2000 for details). This threshold was chosen because the maximum number of false-positive predictions in this analysis is expected to be Np = 14,226 × 10−4 or ∼1.4. In preliminary tests, large numbers of repeats, in particular those containing compositional bias, were generated from overlapping sequences. To exclude such slightly off-diagonal alignments, cases were discarded where the two sequence fragment residue ranges overlapped by >50%.
The aim of this study was to detect hitherto unknown domains and repeats. To establish which of the repeats identified byprospero have been characterized previously, all sequences containing these repeats were searched against the PFAM and SMART databases of HMMs (Bateman et al. 2000; Schultz et al. 2000). Sequences annotated by these resources as containing known repeats were removed. In order that homologous repeats from different fly proteins could be considered together in subsequent analyses, remaining sequences were then clustered using the grouper program of theSEALS package with a default single-linkage clustering cutoff of 50 bits (Walker and Koonin 1997). This generates groups containing sequences that are each aligned by BLAST with at least one other in the group with an alignment score ⩾50 bits. Repeats that could not be clustered, as their alignment scores were <50 bits, were considered separately from groups as “orphan” sequences.
Semi-Automated Analysis
Subsequent analytical steps were undertaken using well-established database searching algorithms. Fully automated analyses were not attempted because computational tools that determine the biological relevance of sequence similarity are not available. For each group a multiple sequence alignment was produced using theClustal-W package (Thompson et al. 1994). All sequences in each group were searched against a nonredundant protein sequence database (nrdb; ftp://ncbi.nlm.nih.gov/pub/blast/db/) using the position-specific and iterative version of BLAST(PSI-BLAST; Altschul et al. 1997) and an E-value inclusion threshold of 2 × 10−3. For those groups for which PSI-BLAST identified additional homologs, a HMM was constructed and compared, using HMMER(http://hmmer.wustl.edu/), against the nrdb, or else a second nrdb (nrdb90; ftp://ftp.ebi.ac.uk/pub/databases/nrdb90/) that contains no pair of sequences with >90% amino acid identity. Sequences identified by these HMMER searches with E<0.1 were considered to be homologous. Additional PSI-BLAST and/orHMMER database searches were initiated using newly identified homologs in an iterative manner until no further homologs were detected. This exhaustive search protocol is similar to that used previously for detecting eukaryotic signaling domains (Ponting et al. 1999).
Determining the extent of domains and repeats often is problematic. Here we have estimated their boundaries by domain architecture analysis. The domain architecture of a protein is defined as the composition and order of its domains and repeats. Repeat lengths were often deduced from instances when they occurred in tandem. In remaining cases, boundaries were assigned by virtue of the presence of neighboring domains or bona fide N- or C-termini, or else, for domains, by detailed consideration of the amino acid conservation of flanking regions of multiple alignments that were extended towards both termini.
Groups were annotated according to experimentally determined molecular functions, or else using resources detecting transmembrane helices (http://www.ch.embnet.org/software/TMPRED_form.html), or protein domains, repeats, and motifs (http://smart.embl-heidelberg.de/ andhttp://www.sanger.ac.uk/Pfam/). For a small minority of groups, protein tertiary structural information was available. This was accessed using Web resources including Scop (http://scop.mrc-lmb.cam.ac.uk/scop/) and PDBsum (http://www.biochem.ucl.ac.uk/bsm/pdbsum/).
The overall flow of the analysis pipeline is illustrated in Figure 1. Multiple alignments and HMMs of domain and repeat families (Table 1) have been added to the SMART resource (Schultz et al. 2000). Hand-curated annotations of groups and orphan sequences are available from: http://www.mrcfgu.ox.ac.uk/ponting/fly_rpts/.
Acknowledgments
C.P.P. thanks Dr. Pat Clissold for assistance in some of the analysis.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL Chris.Ponting{at}anat.ox.ac.uk; FAX 44-1865-272175.
-
Article published on-line before print: Genome Res.,10.1101/gr.198701.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.198701.
-
- Received May 31, 2001.
- Accepted September 10, 2001.
- Cold Spring Harbor Laboratory Press








