
Loss of Heterozygosity Assay for Molecular Detection of Cancer Using Energy-transfer Primers and Capillary Array Electrophoresis
- Igor L. Medintz1,
- Chyi-Chia Richard Lee2,
- Wendy W. Wong1,
- Kristin Pirkola3,
- David Sidransky2, and
- Richard A. Mathies1,4
- 1Department of Chemistry, University of California, Berkeley, California 94720 USA; 2Department of Otolaryngology — Head and Neck Surgery, Division of Head and Neck Cancer Research, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205 USA; 3Molecular Dynamics, Sunnyvale, California 94086 USA
Abstract
Microsatellite DNA loci are useful markers for the detection of loss of heterozygosity (LOH) and microsatellite instability (MI) associated with primary cancers. To carry out large-scale studies of LOH and MI in cancer progression, high-throughput instrumentation and assays with high accuracy and sensitivity need to be validated. DNA was extracted from 26 renal tumor and paired lymphocyte samples and amplified with two-color energy-transfer (ET) fluorescent primers specific for loci associated with cancer-induced chromosomal changes. PCR amplicons were separated on the MegaBACE-1000 96 capillary array electrophoresis (CAE) instrument and analyzed with MegaBACE Genetic Profiler v.1.0 software. Ninety-six separations were achieved in parallel in 75 minutes. Loss of heterozygosity was easily detected in tumor samples as was the gain/loss of microsatellite core repeats. Allelic ratios were determined with a precision of ± 10% or better. Prior analysis of these samples with slab gel electrophoresis and radioisotope labeling had not detected these changes with as much sensitivity or precision. This study establishes the validity of this assay and the MegaBACE instrument for large-scale, high-throughput studies of the molecular genetic changes associated with cancer.
Microsatellite DNA sequences or short tandem repeat (STR) sequences are extremely useful for genetic disease studies, as well as personal identification and gene mapping, due to their high degree of polymorphism and genome-wide distribution (Edwards et al. 1991; Fu et al. 1992; Mao et al. 1996). Loss of heterozygosity (specifically, loss of a microsatellite allele) is a common genetic lesion in many solid neoplasms (Sidransky 1997). Mapping of specific chromosomal regions of LOH and identification of microsatellite expansions and deletions will help identify target genes whose loss, or amplification, is required for cellular transformation (Sidransky 1997; Lasko et al. 1991). The correlation between these molecular genetic changes and tumor progression will also aid in prognosis by determining the possibility of recurrence as well as the choice of treatment.
Previous work has laid a strong foundation for microsatellite analysis of cancer development. Molecular detection of primary bladder cancer, via microsatellite analysis of urine sediment and tumor samples, has been demonstrated (Mao et al. 1996). Recurrence of bladder cancer has also been predicted by microsatellite analysis of urine samples from post-treatment patients (Steiner et al. 1997). However, these assays relied on radioisotope labeling and slab gel technology. To translate this type of assay into a viable clinical tool, large-scale studies of the genetic changes associated with different tumors and different stages of tumor progression need to be undertaken with high-throughput modern instrumentation. As a first step, Wang et al. (1997) validated microsatellite-based cancer detection using prototype capillary array electrophoresis apparatus and energy-transfer fluorescent primers. Di-, tri-, and tetra-nucleotide repeat markers, which have shown expansion, deletion, or LOH associated with primary bladder tumors, were examined on CAE using single- and two-color assays (Wang et al. 1997). The use of energy-transfer dye-labeled PCR primers allowed for the excitation of differentially labeled normal and tumor PCR amplicons at a common laser wavelength, while detecting non-overlapping emissions from the two samples (Wang et al. 1997; Ju et al. 1997).
To further evaluate the potential of LOH and other molecular genetic changes associated with primary tumors as clinical tools, we needed to perform larger scale studies on robust commercial apparatus. To this end, we analyzed 25 renal carcinoma samples along with their paired peripheral normal lymphocyte controls using the MegaBACE capillary array electrophoresis system. Isolated DNA was subjected to amplification at four loci on chromosome 9 within a common deleted region in this type of cancer (Steiner and Sidransky 1996). PCR amplicons from matching tumor and normal samples were separated individually and co-injected to validate measurements taken from co-eluting samples. Resulting electropherograms were analyzed with MegaBACE Genetic Profiler v.1.0 software for molecular size and peak height. Allele ratios were calculated from these peak heights. The results demonstrate that large-scale assaying of microsatellite LOH and MI is easily and rapidly accomplished on the MegaBACE instrument.
RESULTS
One-color STR Analyses
Samples, prepared as described below in the Methods section, underwent a preliminary screening run to determine if the PCR amplification was successful and if samples were homozygous or non-informative (NI) at particular loci (Table1).1 All samples were successfully amplified on the first attempt using the step-down PCR protocol. The amount of PCR product analyzed was adjusted based on the relative fluorescence observed, which overcame any potential sample overloading problems. Panel A of Figure 1 presents a total electropherogram of processed data. A normal blood sample amplified with the ET-ROX D9S747 primer was mixed with TAMRA-labeled sizing standards. Relative fluorescence is plotted against size in bp, as determined from the sizing standards. The peaks around 50 to 100 bp are due to primer and amplification/injection artifacts. Panels B and C show a comparison of this normal sample against a corresponding tumor sample from the same patient. The allelic ratio of the normal sample is 1.12 as compared to 1.89 for the tumor sample. The normalized ratio for this tumor sample is 0.60. A variation of 20% in allelic ratio is considered diagnostically significant with our methodology. The 20% value was determined through a detailed evaluation of the effect of PCR variability and fluctuations related to electrokinetic injection on the sensitivity and specificity of LOH analysis by CE (Wang et al. 1997).
Comparison of Radioactive and ET-CAE Fluorescent LOH Assays
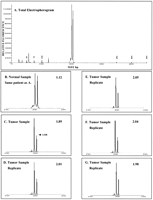
Comparison of normal and tumor samples demonstrating LOH from sample set 10. Samples were amplified at the D9S747 locus with the ET-ROX primer. Relative fluorescence is plotted against the size of the PCR product. (A) Total electropherogram of the normal sample along with the sizing standards. (B,C) Electropherograms from a normal lymphocyte control and a tumor sample from the same patient. (D–G) Replicate injections in multiple capillaries of the tumor sample. The allele ratios are given in the right corner of each panel. See the Methods section for run conditions.
The reproducibility of these CAE separations was examined by performing multiple runs of each PCR sample. Panels D through G present multiple injections of the same tumor sample from Panel C but on different capillaries. The average allelic ratio of the runs in Panels C through G is 1.98 with a standard deviation of 0.06. The mean normalized ratio of the tumor samples against multiple injections of the normal sample was 0.56 with a standard deviation of 0.06. These data confirm that quantitative LOH assays can be performed on the MegaBACE instrument rapidly and precisely.
Two-color STR Analyses
Because LOH assays will ultimately involve large numbers of tumor samples analyzed at multiple STR loci against their corresponding normal lymphocyte controls, two-color, co-injected analysis would be highly desirable. Because it is essential to have labels with as little spectral overlap as possible, ET primers, with a cyanine donor dye and ROX or R110 as the acceptor dyes, were chosen. Both ET primer sets absorb at the common laser excitation wavelength of 488 nm, while ET-ROX emits at 605 nm and ET-R110 at 530 nm (Hung et al. 1996, 1998;Ju et al. 1997).
Figure 2 presents the results of a two-color LOH analysis. Panels A and B show the normal sample amplified at the D9S171 locus, with either ET-ROX (A) or ET-R110 (B), and analyzed in separate capillaries. The ET-ROX is detected in the red channel while the ET-R110 is detected in the blue channel. Panels C and D show the same analysis for the corresponding tumor sample. Panel E presents the results of co-injecting the normal sample amplified with ET-ROX and the tumor with ET-R110, and the converse in Panel F. The normalized allelic ratio for the tumor/normal ET-ROX samples is 0.81, while that of the tumor/normal ET-R110 is 0.79. The mean normalized value from the two-color analysis is 0.80 ± 0.01. The normalized allele ratios for the two-color, mixed tumor/normal samples in panels E and F are 0.76 and 0.84. The mean normalized allelic ratio is 0.80 with a standard deviation of 0.04. This result would place this particular tumor sample at a threshold or borderline LOH value (Wang et al. 1997). These results demonstrate that the sensitivity and precision of this assay on the MegaBACE instrument is not lost when differentially amplified samples are co-injected. The use of different ET dye combinations on the primers leads to slight electrophoretic migration differences of no more than 1 to 1.5 bp (Hung et al. 1996). Also note the presence of stutter peaks that occur from polymerase slippage during amplification of these di-nucleotide repeat STR loci (Wang et al. 1997). The use of the Genetic Profiler software helps overcome confusion from stutter peaks, because the correct allele peaks are easily identified from the sizing function.
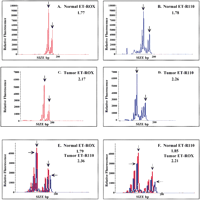
Two-color LOH assays using complex samples (sample set 10). (A,B) Electropherograms from a normal sample amplified at the D9S171 locus with both ET-ROX (red) and ET-R110 (blue) primers. (C,D) Electropherograms from the matching tumor samples with both primer sets. (E,F) The resulting electropherograms from co-injected two-color samples. The numbers given at the right of each panel are the allelic ratios. The arrows indicate the peaks used for allele ratio determination.
The use of normalized allelic ratios from tumor and serum control samples is of critical importance in these assays, which are quite sensitive to changes in peak morphology. Recent evidence suggests that there is a significant diversity of allele ratios and morphology when STR loci are typed via PCR and electrophoresis. Trends seem to suggest that the further STR alleles are separated in size, the greater the size difference in relative peak heights. Additionally, it appears that the stutter band corresponding to the larger allele increases in intensity as a function of increased size (David Shen, Molecular Dynamics, unpubl.). The use of normalized allelic ratios significantly reduces the ambiguity that can be caused by this problem.
Figure 3 presents representative results where the smaller allele in a tumor sample is completely lost. Panel A shows the normal sample amplified at the D9S171 locus with the ET-R110 primer, while Panel B shows the electropherogram from the corresponding tumor sample. The 176 bp smaller allele is completely lost in the tumor sample. Panel C presents the electropherogram from the co-injection of both samples.
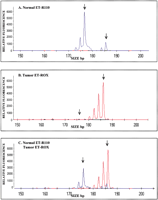
Two-color electropherograms of D9S171 loci amplicons showing complete loss of heterozygosity (sample set 22). (A,B) Normal sample amplified with the ET-R110 (blue) labeled primer and the tumor sample amplified with the ET-ROX (red) labeled primer, respectively. (C) Co-injected mixed sample. The arrows indicate the two alleles being compared. The slight difference in intensity is caused by dilution of one sample into another.
Figure 4 presents the results from two separate tumor samples where microsatellite instability is demonstrated by the expansion and deletion of STR repeats, which change the size of the alleles. Both sets of samples were amplified at the D9S162 locus. In Panel A, the larger allele gains five di-nucleotide repeats and increases in size by 10 bp, while in Panel B the converse is demonstrated. The larger allele in the tumor sample presented in Panel B loses five di-nucleotide repeats, and thus decreases in size by 10 bp. The sizing function of the software is important here because the size and number of di-nucleotide repeats gained or lost are easily determined.
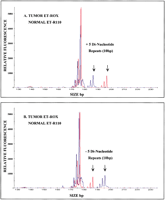
Two-color electropherograms from the D9S162 loci demonstrating nucleotide repeat expansion or deletion associated with microsatellite instability (MI). (A) Ten-bp size increase caused by an expansion of five di-nucleotide repeats in the ET-ROX (red) amplified tumor sample as compared to the ET-R110 (blue) amplified normal sample (sample set 5). (B) Ten-bp size loss caused by the deletion of five di-nucleotide repeats in the ET-ROX (red) tumor sample as compared to the ET-R110 (blue) normal sample (sample set 22).
Comparison of Radioisotope to Fluorescent LOH Assay
Table 1 summarizes the fluorescent ET-CAE LOH analysis of the 26 sets of renal carcinomas compared to radioisotope slab gel-based LOH analysis carried out on these same samples. The number presented in the ratio column is the normalized allelic ratio for that particular tumor/serum sample at that locus. The fluorescent technique is able to detect LOH in a significantly higher number of samples than previously detected by the radioisotope LOH assay. This is especially evident at the D9S171 locus where an additional eight samples had significant LOH when tested with this improved assay using the 20% variation criteria (Wang et al. 1997). Some of these additional samples have positive but borderline LOH values, which are expected when using an assay with higher sensitivity. For example, sample set 25 demonstrated LOH of the lighter allele at a borderline value (0.77). Similar examples of borderline LOH values are found at the other three loci as well.
An inability to resolve heterozygous samples that appear to be closely related in size (2 bp difference) was noted for sample set 24 at the D9S162 locus. The use of alternative matrix chemistry that has a higher concentration of denaturing agents, plus the use of a higher formamide concentration in the sample and longer denaturing times, should address this problem.
It appears that the inconsistencies between the radioisotope assay (RA) and the fluorescent assay can be classified into three types. Examples of the first type of inconsistency can be found at the D9S747 locus for sample sets 6 and 11. Here, the RA indicated LOH, whereas the fluorescent normalized ratio was not significant. Examples of the second type of inconsistency are found throughout this study, where the fluorescent assay indicated a significant normalized ratio variation, while the RA did not. The third type of inconsistency is typified by D9S747, set 26, and D9S171, set 6. Here the fluorescence assay indicates significant LOH while the RA assay indicates a non-informative locus. Additionally, examples of microsatellite instability are detected by the ET-CAE LOH assay that were not detected by the radioisotope assay (D9S162 locus, sample sets 5 and 22). These differences may arise from the fact that the radioisotope assay has a large amount of intrinsic exposure variation. The fact that larger alleles may have a much lower intensity relative to the smaller allele may also lead to misinterpretation of autorads. Consistent use of normalized ratios and the sizing function of the Gene Profiler program can alleviate this problem when using the fluorescent assay.
DISCUSSION
We have investigated the use of the MegaBACE-1000 CAE system for detecting LOH in clear cell renal carcinoma samples. Using one- and two-color fluorescent ET labeling, LOH allelic ratios are determined (with reproducibility and accuracy) with a variation of less than ± 10%. A direct comparison of this assay with a radioisotope-labeling assay clearly demonstrated the higher sensitivity and specificity of our approach for LOH analysis. The values for RA analysis presented in Table 1 were generated as part of a diagnostic study (Eisenberger et al. 1999). The fluorescent assay detected LOH in a significantly higher number of samples, along with examples of microsatellite instability that went undetected by RA analysis alone. Indeed, this fluorescent assay found significant LOH in a sample set that had been characterized as non-informative or homozygous by RA (see sample set 26 at the D9S747 locus). Increased sensitivity may result in a number of added benefits. These include a reduction in the number of markers to be tested if the loci already selected are likely to be more informative. Increased specificity could also circumvent the need to select out non-neoplastic cells for enrichment. By obtaining information of higher sensitivity at specific loci, the assay would also promote better prognostic/predictive outcomes.
A variety of additional benefits are clearly evident from this preliminary study. The use of Genetic Profiler software to size all pertinent alleles allows instant identification of real allele peaks as opposed to PCR stutter bands. This will be extremely important in cases of extreme LOH where an allele may be entirely lost (Fig. 3). Using this software for analysis also facilitates rapid and accurate determinations of allele size shifts caused by nucleotide instability. Traditional methods of sizing radiolabeled alleles present on exposed X-ray films or captured gel images are cumbersome, time consuming, and may be error-prone.
Additionally, Genetic Profiler software allows instant archiving and identification of alleles in samples and composite statistical analysis of data across an entire project that may consist of many hundreds of individual runs. This will allow the easy elucidation of particular allelic changes or instabilities at different loci that may be correlated with particular cancers or stages of cancer.
Finally, the ability to perform 96 assays in parallel in a 75 minute time period, with an approximate turnaround time of one hour, is especially suited to large-scale, high-throughput studies of LOH across many loci and multiple types of cancer. The analysis of the vast amount of data retrieved from such a study will allow for a better understanding of cancer development, and lead to better clinical tools for the early detection, diagnosis, prognosis, and possibly, directed treatment of cancer.
METHODS
Samples
DNA from patients was isolated from 25 matched sets of renal carcinomas and paired peripheral blood lymphocyte controls. These samples had previously undergone analysis for LOH using radioisotope labeling and slab gel electrophoresis (Mao et al. 1996; Steiner et al. 1997; Eisenberger et al. 1999).
PCR Primers and Amplification of STR Loci
Four sets of two-color, energy-transfer fluorescent PCR primers were synthesized according to the methodology of Hung et al. (1996,1998) and Ju et al. (1995). The labeled primers were all conjugated with a common fluorescent donor cyanine dye (CYA), 3-(ɛ-carboxypentyl)-3′-ethyl-5-5′-dimethyloxacarbocyanine, at the 5′ end of the primer, and the acceptor dyes were conjugated onto a modified T nucleotide within the sequence. Acceptor dyes were either 6-carboxy-X-rhodamine (ROX) or 5- and 6-carboxyrhodamine-110 (R110) (Molecular Probes, Eugene OR, USA). Primer sequences and microsatellite loci characteristics are given in Table2. Amplification utilized a modified step-down protocol (Hecker et al. 1996). PCR samples underwent initial denaturation at 95° C for three minutes, followed by three cycles of denaturing at 95° C for 30 seconds, annealing at 64° C for 1 minute and extending at 72° C for 1 minute. Annealing temperatures were dropped 2° C for subsequent three cycle repeats until 52° C. Cycling was completed with 35 cycles that had an annealing temperature of 50° C, followed by 72° C for 7 minutes and a 4° C hold.
Sequences and Characteristics of ET Primers
Sample Preparation and Sizing Standards
PCR amplicons were dialyzed against 0.1 X TE buffer for at least 20 minutes. Samples were then diluted between 5- and 30-fold in distilled/deionized H20, and 1 μL of diluted sample was mixed with 4–5 μL of 75% deionized formamide containing dye-labeled sizing markers (10 fM). The sizing standard consisted of six fragments (50, 75, 100, 350, 450, and 500 bp) covalently labeled with TAMRA, N, N, N′, N′,-tetramethyl-6-carboxyrhodamine (Bio Ventures Inc., Murfreesboro, TN USA). Samples were placed in a 96-well microtiter injection plate and centrifuged for 5 seconds to remove air bubbles. Fewer than 10 minutes before injection, the samples were denatured at 95° C for at least 4 minutes and immediately placed on ice.
Instrumentation
A MegaBACE-1000 capillary array electrophoresis system (Molecular Dynamics, Sunnyvale, CA USA) was utilized in this study. This CAE system contains 96 capillaries, each with a 40 cm length (Bashkin et al. 1996a,b). Long Read linear polyacrylamide matrix, LPA, containing 7M urea in a Tris/Taps buffer was obtained from Amersham/Pharmacia Biotech, Piscataway, NJ USA. The matrix was introduced into the capillaries at 400 psi for 200 seconds and then allowed to relax for 20 minutes, followed by pre-electrophoresis for 5 minutes at 10 kilovolts (kV). Denatured samples were introduced into the capillaries by electrokinetic injection for 45 seconds at 3 kV and then electrophoresed at 10 kV for 75 minutes. Fresh matrix was introduced into the capillaries between each run.
Data Analysis
Data were analyzed with the MegaBACE Genetic Profiler v.1.0 software developed at Molecular Dynamics. The raw run image data were imported into the Genetic Profiler program where they underwent background subtraction using a spectral separation or cross-talk matrix. Unknown samples were sized against the known TAMRA-labeled size markers included in each capillary using a third-order local algorithm. Peak height or peak intensity (relative fluorescence) data were also generated in this step. Due to limited tumor sample DNA, some of the IFN-A analysis was carried out in only one color as opposed to the full two-color analysis.
Calculation of Allelic Ratios
Allelic ratios are calculated as the peak height of the smaller/lighter peak divided by the peak height of the larger/heavier peak. Empirically, peak heights appear to be superior to the use of peak areas, in agreement with the findings of others (Paulson et al. 1999). Amplification of DNA samples derived from lymphocytic blood cells provided an allelic control. To estimate the degree of LOH, normalized ratios are calculated as (T1/T2)/(N1/N2) where T1 and N1 are the peak heights of the lighter alleles and T2 and N2 are the peak heights of the heavier alleles (Wang et al. 1997). For the ratios presented in Table1, the reciprocal value was used for ratios greater than 1.
Acknowledgments
This work was supported by NIH Program Project grant PO1 CA77664, awarded to Johns Hopkins University and to the University of California at Berkeley. Additionally, the authors thank Dr. Mark Lewis, Dr. Melanie Mahtani, and David Shen of Molecular Dynamics for assistance in using the MegaBACE-1000 system.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL Rich{at}zinc.cchem.berkeley.edu; FAX 510-642-3599.
-
- Received October 12, 1999.
- Accepted May 18, 2000.
- Cold Spring Harbor Laboratory Press








