
Abstract
Multiple sequence alignment (MSA) is a critical step in the study of protein sequence and function. Typically, MSA algorithms progressively align pairs of sequences and combine these alignments with the aid of a guide tree. These alignment algorithms use scoring systems based on substitution matrices to measure amino acid similarities. Although successful, standard methods struggle on sets of proteins with low sequence identity: the so-called twilight zone of protein alignment. For these difficult cases, another source of information is needed. Protein language models are a powerful new approach that leverages massive sequence data sets to produce high-dimensional contextual embeddings for each amino acid in a sequence. These embeddings have been shown to reflect physicochemical and higher-order structural and functional attributes of amino acids within proteins. Here, we present a novel approach to MSA, based on clustering and ordering amino acid contextual embeddings. Our method for aligning semantically consistent groups of proteins circumvents the need for many standard components of MSA algorithms, avoiding initial guide tree construction, intermediate pairwise alignments, gap penalties, and substitution matrices. The added information from contextual embeddings leads to higher accuracy alignments for structurally similar proteins with low amino-acid similarity. We anticipate that protein language models will become a fundamental component of the next generation of algorithms for generating MSAs.
Multiple sequence alignments (MSAs) underlie much of sequence analysis in computational biology. Protein MSAs are the main input to algorithms for reconstructing gene and species trees (Felsenstein 2004); for identifying conserved (Capra and Singh 2007), co-evolving (Marks et al. 2011), specificity-determining (Capra and Singh 2008), and positively selected sites (Miyata and Yasunaga 1980); for building protein motif and domain representations (Sonnhammer et al. 1998); and for predicting de novo protein structural features (Rost and Sander 1994) and full protein structure prediction (Jumper et al. 2021). Highly accurate MSAs are necessary, as misaligned amino acids can lead to incorrect inferences for this wide range of downstream tasks. However, when the average sequence identity among the sequences to be aligned is low, most MSA methods struggle to obtain high-quality alignments (Nute et al. 2019); sequence identity below 20%–35% has been termed the “twilight zone” of alignment (Rost 1999).
Over the past 40 years, numerous algorithms for obtaining MSAs have been developed and refined, including CLUSTAL (Sievers et al. 2011), MAFFT (Katoh and Standley 2013), Muscle (Edgar 2004), T-Coffee (Notredame et al. 2000), ProbCons (Do et al. 2005), Saté (Liu et al. 2012), and PASTA (Mirarab et al. 2015), among others. The predominant approach to align multiple sequences is to first align pairs of sequences and then progressively combine these subalignments into larger alignments in an order determined by a guide tree (Feng and Doolittle 1987). Pairwise alignment algorithms (Needleman and Wunsch 1970; Smith and Waterman 1981) uncover a highest scoring alignment between two sequences when given a similarity measure between all pairs of amino acids and a gap penalty function. Similarities between each pair of the 20 amino acids are typically based upon substitution matrices that encapsulate the frequency with which amino acid pairs are observed in equivalent positions across large data sets of alignments of homologous sequences (Dayhoff et al. 1978; Altschul 1991; Henikoff and Henikoff 1992). Although in theory optimal pairwise alignment algorithms can be adapted to multiple sequences, the runtime would be exponential in the number of sequences, and further, it is not clear how to score multiple amino acids in a single aligned column. Instead, guide tree–based MSA approaches are heuristics that optimally combine pairs of alignments in which the similarity of a pair of columns in two different alignments is computed based on substitution matrix scores of the amino acids within the columns. Alternatively, consistency-based progressive MSA approaches use similarity scores that consider how frequently pairs of amino acids in two different sequences are aligned to the same amino acid position in other sequences when considering optimal pairwise alignments (Notredame et al. 1998, 2000).
Here, we consider a new approach to determining the similarities between amino acids across proteins, based on protein language models (Rao et al. 2019; Bepler and Berger 2021; Rives et al. 2021; Chowdhury et al. 2022; Elnaggar et al. 2022). Protein language models are “self-supervised” deep learning language models that have been pretrained on large compendia of protein sequences, generally trained to predict “masked” amino acids within input protein sequences based on the rest of the sequence. Once trained, these models embed each amino acid within a protein sequence as a high-dimensional vector, and importantly, these vectors capture the sequence “context” of each amino acid. Machine learning models using these embeddings have been successfully trained to predict many protein structural and functional features, including conservation (Marquet et al. 2022), ligand binding (Littmann et al. 2021), homology (Rives et al. 2021; Heinzinger et al. 2022), and subcellular localization (Stärk et al. 2021). Because amino acids at equivalent positions in two different protein sequences play the same structural and functional role, we reasoned that clustering amino acid context vectors across sequences could be the basis of a new approach for aligning sequences. Although recently neural network machinery has been leveraged to perform dynamic program-based alignments (Morton et al. 2020; Petti et al. 2023), our approach is, to the best of our knowledge, the first that uses protein sequence embeddings to identify analogous positions across protein sequences in order to determine MSAs. Our MSA approach is based on clustering the amino acid contextual vectors produced by protein language models and then using graph-theoretic approaches to determine a consistent ordering of MSA columns. Our method, vector-clustering Multiple Sequence Alignment (vcMSA), is a true multiple sequence aligner that aligns multiple sequences at once instead of progressively integrating pairwise alignments. Our core methodology diverges from standard MSA methods in that it avoids substitution matrices and gap penalties, and in most cases, does not use guide tree construction. In proof-of-concept testing, we aim to establish the power of leveraging protein language models for building MSAs, particularly for low identity sequence families that current methods have the most difficulty on.
Methods
Overview
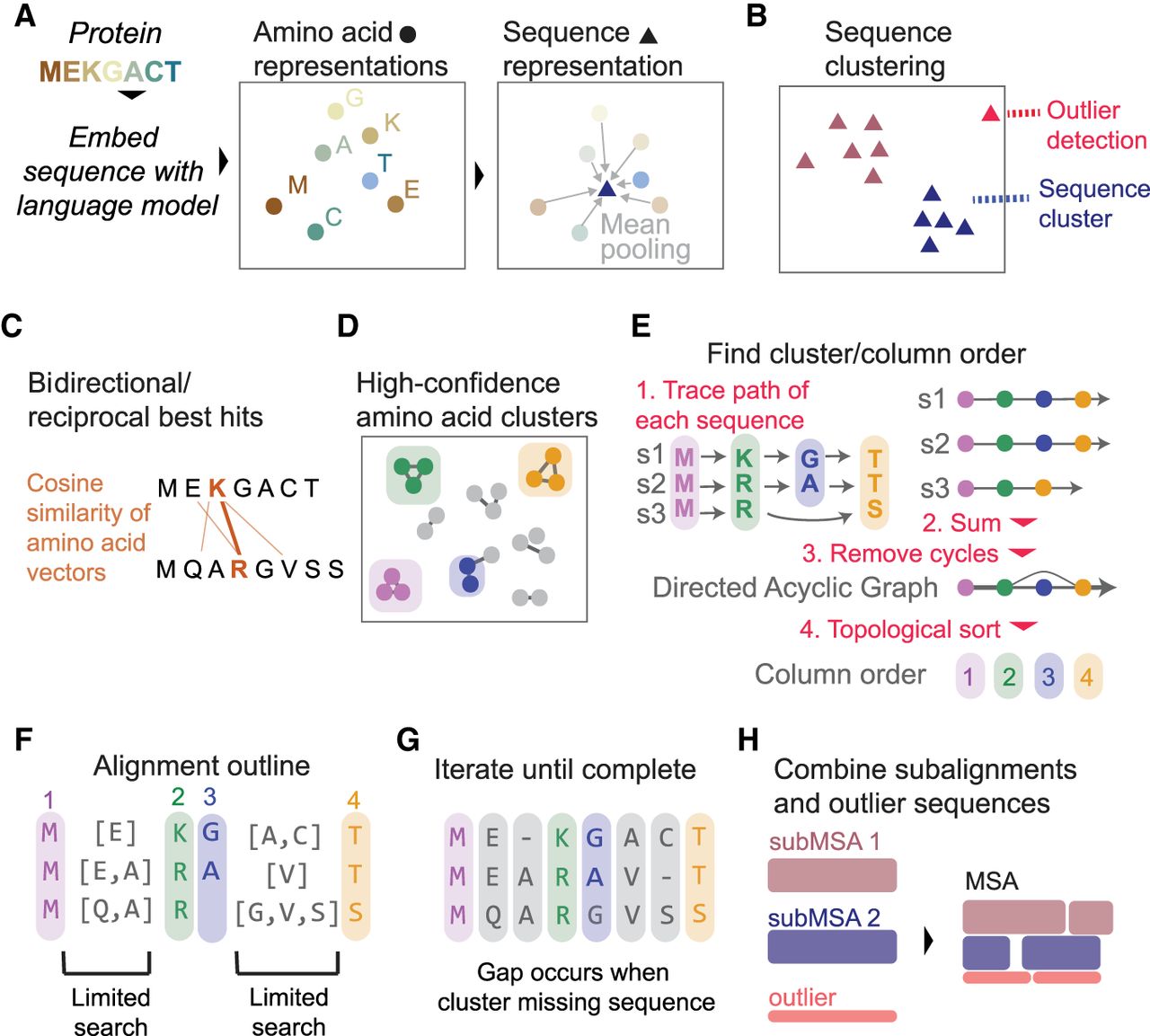
A brief overview of our algorithm is as follows. First, for all protein sequences, we obtain per-position and sequence-level embeddings via a protein language model (Fig. 1A). Second, we cluster protein sequences via the sequence-level embeddings to uncover sets of sequence-similar proteins (Fig. 1B). Third, within each cluster of sequences, we measure the similarities of amino acid vectors and, for each amino acid, find the one that is most similar to it in each of the other sequences (Fig. 1C) and filter to amino acid pairings that are reciprocal best hits (RBHs) of each other. Fourth, we build an RBH network and cluster the network to find “guidepost” amino acid positions in different sequences that are clearly aligned to each other (Fig. 1D). Fifth, we order the clusters into columns of the MSA by constructing a directed acyclic graph (DAG) based on sequential positions within each protein sequence and performing a topological sort (Fig. 1E). Sixth, we use guidepost clusters to limit the scope of searches for the remaining unplaced amino acids (Fig. 1F). Seventh, we continue creating guideposts and assigning amino acids to columns until all amino acids are placed in the alignment (Fig. 1G). Finally, we combine subalignments from the sequence clusters into the final MSA (Fig. 1H). Further details about each of these steps are provided below.
Overview of vcMSA algorithm. (A) Proteins are embedded using a protein language model to produce vector representations of each amino acid, and the mean of these amino acid embeddings is taken to produce a sequence-level representation. (B) We cluster sequence representations and detect outlier sequences. (C) For each sequence cluster, we determine bidirectional/reciprocal best hits (RBHs) of cosine similarity between pairs of amino acids in different sequences. (D) From a network built from RBHs, we determine confident clusters of amino acids, corresponding to columns in the MSA. (E) To determine column order, we trace the path of each sequence through clusters and combine all paths into one network, taking edge weights from the number of sequences that traverse between the pairs of clusters. We trim any clusters that cause cycles and use a topological sort of the resulting directed acyclic graph to find column order. (F) Clusters/columns limit scope of search for unplaced amino acids. (G) We iterate limited searches until all amino acids are placed. Gaps in the alignment occur when a cluster does not contain an amino acid from a sequence. (H) We combine alignments from each sequence cluster and outliers in the final output MSA.
Embedding generation
For each protein sequence, we use the encoder portion of the ProtT5-XL-UniRef50 language model to generate sequence embeddings (Elnaggar et al. 2022). T5 models undergo self-supervised training on a large corpus of text, in this case over 11 million protein sequences in the UniRef50 data set (Elnaggar et al. 2022). For each amino acid in a sequence, the ProtT5-XL-UniRef50 model produces one embedding vector of length 1024 for each of its 24 encoder layers. We choose to use embeddings from the final 16 layers, giving us a final vector representation of each amino acid with dimension 16,384. For each sequence, we average all of the vector embeddings for each of its amino acids to obtain a sequence-level embedding. We additionally add padding to sequences, where we first add 10 X's to the start and end of each sequence before embedding (Supplemental Fig. 1A).
Sequence clustering
We find that vcMSA performs best on semantically consistent sets of sequences and that performance is degraded when outlier sequences are included. Therefore, we first cluster input sequences based on sequence-level embeddings to obtain groups of similar sequences and detect outliers (Supplemental Methods). We then apply the remaining steps of vcMSA to each cluster of sequences individually. Left out sequences and alignments for clusters are merged into a full alignment as described below.
Amino acid similarity across sequences
The contextual similarity between a pair of amino acids is defined as the cosine similarity between their embedding vectors. To reduce sequence-specific effects and increase the overall similarity between different sequences, we first apply “batch correction” to amino acid embeddings in the same sequence cluster, considering each sequence as analogous to a batch (Supplemental Fig. 1C; Supplemental Methods). For each amino acid in each sequence, we next find the most similar amino acid in each of the other sequences by considering cosine similarities between their embeddings. We use Facebook's fast AI similarity search (faiss) library to perform nearest neighbor searches for all amino acids (Johnson et al. 2021). For each pair of sequences, the set of pairwise similarities between amino acids between different sequences is filtered to only RBHs, for which the best match to a query amino acid in the target sequence must also have its highest score in the query sequence be the query amino acid (Fig. 1C). For each pair of sequences, we construct a bipartite graph composed of nodes for each amino acid in the two sequences, with an edge for each RBH identified between amino acids in the two sequences. We will keep a largest set of these RBHs that are “consistent” with each other in a pairwise alignment using a maximum noncrossing matching (MNCM) formulation (Malucelli et al. 1993). In particular, if the ith and jth amino acids in the first sequence (we assume without loss of generality that i < j) have as their RBHs the kth and lth amino acids, respectively, in the second sequence, then this pair of RBHs is consistent only if k < l; that is, if k < l, it is possible for a pairwise alignment between these two sequences to simultaneously align the ith and jth amino acids in the first sequence with the kth and lth amino acids in the second sequence. If two pairs of RBHs are not consistent with each other, we say that the edges that correspond to them “intersect,” as they would if the nodes were arranged in two columns (one for each sequence) in the order in which they appear in the sequences. We define a noncrossing matching to be a selection of edges such that no two edges intersect, and an MNCM to be the largest possible set of such edges. In the case that there are multiple MNCMs, we select the MNCM that has the highest average cosine similarity score between all RBHs in the matching. We remove from consideration any RBHs that are not part of this MNCM; in this manner, we obtain a filtered set of RBHs across all pairs of sequences. MNCM performs well in preserving RBHs corresponding to correctly aligned amino acid pairs, while removing false-positive matches (Supplemental Fig. 2).
Amino acid clustering
We build a network in which there is a node for each amino acid in each sequence, and there is an edge between two nodes if the corresponding amino acids are RBHs included in the filtered set of RBHs. We use this network of RBHs in order to cluster sets of amino acids that align to each other with high confidence (Fig. 1D; Supplemental Methods). Amino acids within a cluster will comprise a column in the MSA. We term these aligned positions guideposts and will use these guidepost columns to limit the scope of future searches for amino acids that align to each other (Fig. 1F). Our guidepost columns are similar to user-defined or automatically inferred “anchor points” that have been used in the past to constrain traditional MSA approaches (Morgenstern et al. 2006; Pitschi et al. 2010).
Column order determination
Each cluster corresponds to a column of the MSA; however, these columns must be placed in the correct order in the alignment that is being built. To do this, we will first build a graph based on the correct ordering of amino acids in each protein sequence and then prune this graph to be a DAG and perform a topological sort of this graph (Fig. 1E). In particular, we will introduce a node for each cluster. Next, for clusters A and B, if there exists amino acids u ∈ A and v ∈ B where u and v are in the same sequence, u is before v in the sequence, and v is the first clustered amino acid after u in the sequence, then we will add a directed edge from u to v. This edge will be weighted by the number of sequences in which such u and v exist. Out-of-order amino acids, in which an amino acid is placed in a cluster that occurs too late or too early in the MSA, will cause cycles in this directed graph. To remove these out-of-order amino acids, we detect feedback arc sets (Eades et al. 1993; Csardi and Nepusz 2006), finding edges to remove to obtain an acyclic graph and prioritizing edges with low weight for removal. Because feedback arc set detection is NP-complete, we use the linear time heuristic approach of Eades et al. (1993). Feedback arc set detection has been previously used in MSA to enforce order consistency for multiple local areas of alignment (Pitschi et al. 2010).
For each node-pair of each feedback arc, we discard the node/cluster ID with lowest total edge weight and finally confirm that the trimmed network is a DAG. We then perform a topological sort of the graph to obtain an ordered path that visits every node (cluster ID). This sequence of cluster IDs is the order of columns in the MSA.
Limited scope searches
At this stage, we have a subset of guidepost columns in the alignment, as well as sets of amino acids that have not been assigned to any column (Fig. 1F). If an unassigned amino acid occurs after an amino acid that has been assigned to column p and before an amino acid that has been assigned to column q, we only need to consider amino acids from other sequences that also fall between p and q as candidate matches.
We repeatedly alternate two phases of adding amino acids into the alignment until there are no remaining unplaced amino acids with cosine similarity > 0.1 to any potential match amino acid in the scope. Our two phases are (1) create new columns by amino acid clustering, as described above, among all amino acids also falling between the appropriate guideposts and (2) add amino acids into existing columns by best match score (Supplemental Methods). Existing columns can only be modified during the second phase with the addition of new members. In early iterations following clustering stages, we remove clusters in which any member of the cluster is “stranded,” meaning that neither of its previous or next amino acids is found in the previous and next aligned column/cluster. This quality-control step prevents early errors in which an amino acid is placed in the wrong cluster. For the final stage, amino acids with no matches to any clusters or other amino acids in the scope are placed in their own cluster of size one.
Gaps
Our method does not require parameters related to setting gaps. Once all amino acids are assigned to columns, we lay out the final alignment (Fig. 1G). A column that has one amino acid from every sequence has no gaps. If a column does not contain an amino acid from a particular sequence, a gap is left for that sequence in that column. Our algorithm therefore does not need to use gap opening or gap extension parameters, which are required by other methods.
Merging excess partitions over neighboring columns
We observe cases in which two adjacent columns contain amino acids from a mutually exclusive set of sequences. We detect and merge these paired columns using a low threshold cosine similarity score of 0.1 between amino acids in adjacent clusters, on the basis that independent insertions of one amino acid at the site are biologically unlikely.
Benchmarking
We use the QuanTest2 (Sievers and Higgins 2020) reference alignment data set, which consists of 151 protein sets, each composed of 1000 proteins. The QuanTest2 alignment set is a subset of the HOMSTRAD (Stebbings and Mizuguchi 2004) database of curated structure alignments. We use HOMSTRAD alignments that are not included in the QuanTest2 set to evaluate correspondence between RBH pairs of amino acids and aligned positions. We modified the nextflow pipeline nf-benchmark (Garriga et al. 2019) to wrap our scripts and manage comparison to other methods and evaluation on the QuanTest2 set of alignments.
We compare our method to Clustal Omega (Sievers et al. 2011), MAFFT-FFTNS1, MAFFT-GINSI, MAFFT-LINSI (Katoh and Standley 2013), MUSCLE (Edgar 2004), ProbCons (Do et al. 2005), T-Coffee (Notredame et al. 2000), UPP (Nguyen et al. 2015), and FAMSA (Deorowicz et al. 2016) and build initial guide trees for all methods except UPP with MAFFT-PartTree (Katoh and Toh 2007).
We use the same default parameters for other algorithms as in the supplemental information of Garriga et al. (2019). As with previous work, alignment performance is evaluated using gold-standard reference alignments of the first three proteins in each FASTA file (Garriga et al. 2019). For the proof-of-concept testing described here, we created smaller sets of proteins to align consisting of 20 sequences, for which we select the first three sequences from each FASTA file, as well as 17 randomly selected protein sequences. We additionally remove four protein sets with a more than fourfold difference in length between the three reference sequences and the 17 random sequences, leaving a benchmarking data set of 147 protein sets. We calculate sequence identity for each gold-standard alignment by recording the mean proportion of times each aligned position in the pairs of sequences matches exactly. For sequence identity bins, identities <0.2 fall in bin 0.1, <0.3 fall in bin 0.2, and so on. For runtime measurements, we also create larger sets of proteins consisting of 50, 100, and 200 sequences, where we select the first three sequences and then 47, 97, and 197 sequences, respectively. For evaluating against structured portions of the alignment, we filter down gold-standard alignments to the subset of amino acids with DSSP structure codes H, G, I, E, and B (Kabsch and Sander 1983).
Evaluation score
We use the total column score calculated with the aln_compare plugin from T-Coffee to score sequence alignments against the gold-standard alignments. This score is the percentage of columns in the output alignment that fully match columns in the reference gold-standard alignment.
Speed
On an NVIDIA A100 GPU, initial protein embedding of 20 sequences and indexing of embeddings with faiss take, on average, 22.6 sec, and 100 sequence takes, on average, 31.3 sec, including loading the model. On a CPU configuration, protein embedding is substantially slower, taking 2–5 min to embed 20 sequences, depending on the length of the sequences and CPU memory. The time to produce an alignment depends on the number of sequences in the alignment and on the number of iterations required to place all amino acids (Supplemental Fig. 3A). After embedding, an alignment of 20 sequences when run on an NVIDIA A100 GPU (for similarity searches on the faiss index) and a 2.6-GHz AMD EPYC Rome CPU (remaining steps) with 64 GB of memory takes a median of 1.2 min, for 50 sequences a median of 6.1 min, for 100 sequences a median of 22.6 min, and for 200 sequences a median of 82.9 min.
For comparison, on these data sets, T-Coffee is about five to 10 times faster when run on a 2.6-GHz AMD EPYC Rome CPU with 64 GB of memory. Not including time to build trees, as this varies highly by the tree building method, in our benchmarking, T-Coffee takes a median of 0.2 min for 20 sequences, a median of 1.47 min for 50 sequences, a median of 5.8 min for 100 sequences, and a median of 26.1 min for 200 sequences (Supplemental Fig. 3B). As our implementation of the vcMSA is a proof of concept, we expect the speed of producing alignments to fall substantially with program optimization.
Merging subalignments and excluded sequences
Thirteen of 147 of our QuanTest2 20 protein sets contain more than one cluster of sequences, and 59/147 contain at least one outlier sequence. For these 63 protein sets, we must combine vsMSA produced subalignments and excluded sequences into a complete alignment containing all sequences (Fig. 1H). For this step, we use MAFFT-LINSI merge (Katoh and Standley 2013), which uses our sets of prealigned sequences as internal nodes of a guide tree constructed by UPGMA, using the command mafft ‐‐clustalout ‐‐merge key_table ‐‐auto sub.alns > merged.aln. For the remaining 84 protein sets, no merging of alignments is necessary. Although in a typical use case, outlier sequences would be removed, we include them for compatibility with benchmark protein families.
Results
RBHs between amino acid representations are a solid foundation for MSA
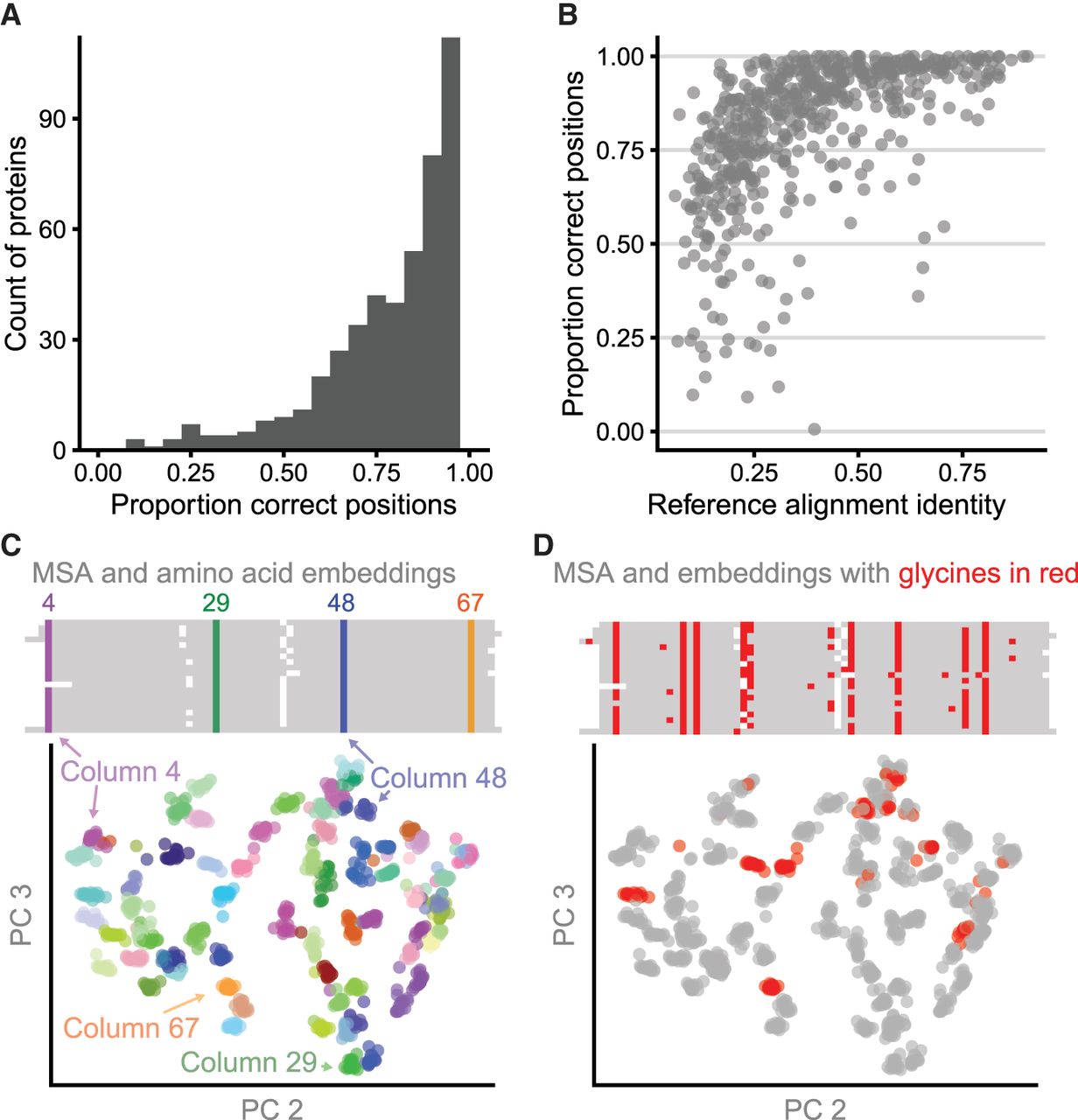
To show that RBHs between amino acid embeddings in two different sequences accurately reflect aligned amino acids, we measured the proportion of aligned positions in 562 pairs of sequences from the HOMSTRAD gold-standard structural alignment database (Stebbings and Mizuguchi 2004) that are RBHs when comparing amino acid embeddings. For 93% of gold-standard protein alignments, at least 50% of aligned columns are additionally RBHs between amino acid embeddings even after filtering RBHs based on uncovering a MNCM (Fig. 2A). The correspondence between RBHs and aligned columns is roughly dependent on sequence identity of the alignment (Fig. 2B). Thus, protein language models are highly effective in identifying analogous positions across protein sequences. These RBHs are the main input to the vcMSA algorithm.
RBHs and clustered amino acid representations reflect aligned columns. (A) For 562 pairs of aligned proteins from the HOMSTRAD database, the MNCMs of RBHs between vector representations frequently correspond to correctly aligned positions. For each gold-standard alignment, we compute the fraction of aligned gold-standard positions that are additionally RBHs after MNCM filtering for that protein pair and display these results as a histogram. (B) The proportion of columns in the gold-standard pairwise alignments that are in the MNCM of RBHs is related to sequence identity of the reference alignment. (C) Principal components 2 and 3 for amino acid vector representations of 20 cold-shock proteins from the csp protein family. Amino acid representations (below) are colored by their corresponding column in the multiple sequence alignment (above). (D) Identical amino acids in different columns are distinguishable from each other. All glycines in the amino acid PCA plot and the multiple sequence alignment are colored red.
Clusters of amino acid representations correspond to columns in the MSA
When amino acid representations in a PCA are colored by their corresponding column in the MSA, it can be seen that amino acids in the same aligned column cluster together (Fig. 2C). We show this correspondence with 20 sequences from the csp protein family using their HOMSTRAD alignment. One initial concern about using amino acid representations for MSA was that the representations of the same amino acid (e.g., glycine) would be too similar wherever that amino acid occurs in the sequence. Instead we find that representations of the same amino acids are distinguishable across the alignment (Fig. 2D).
vcMSA produces more accurate alignments than other algorithms, particularly at low sequence identity
We benchmarked vcMSA on 147 protein alignments consisting of 20 sequences each from the QuanTest2 data set and compared our results to seven alignment algorithms (Fig. 3A). As the metric to evaluate alignment accuracy, we chose total column score, which measures the number of columns that match the gold-standard alignment, owing to its high dynamic range relative to other metrics. vcMSA generally matches or exceeds the alignment accuracy of other algorithms and performs especially well on the lowest sequence similarity proteins in the benchmark. For example, vcMSA is able to align the Asp_Glu_race_D family with a total column score of 63.9, whereas the maximum score of other alignment methods is 0.9.
vcMSA alignments are frequently more accurate than previous methods, particularly for alignments with low levels of sequence identity. We benchmarked our implementation of vcMSA on sets of 20 sequences from each of 147 gold-standard multiple sequence alignments from the QuanTest2 data set. (A) We compare our method to eight state-of-the-art algorithms for sequence alignment. Each point corresponds to one protein MSA and is colored by its sequence identity. Each panel is a tilted scatterplot of relative performance of each other algorithm tested against vcMSA by total column score of produced alignments. Points to the right of the vertical dashed line are alignments where vcMSA outperformed the other method. The number of alignments to the left or right of the line is in gold. vcMSA performs better on more test sets than all other tested algorithms. The point lying on the right axis line is the Asp_Glu_race_D alignment, which all other algorithms perform poorly on (TC score ∼0). (B) We compare the median total column score of vcMSA alignments to MAFFT-GINSI and Clustal Omega alignments, grouping alignments by sequence identity bins. We observe increased scores for vcMSA at low sequence identities. (C) We show median total column scores for structured portions and full gold-standard reference alignments for all algorithms, separated by sequence identity bin.
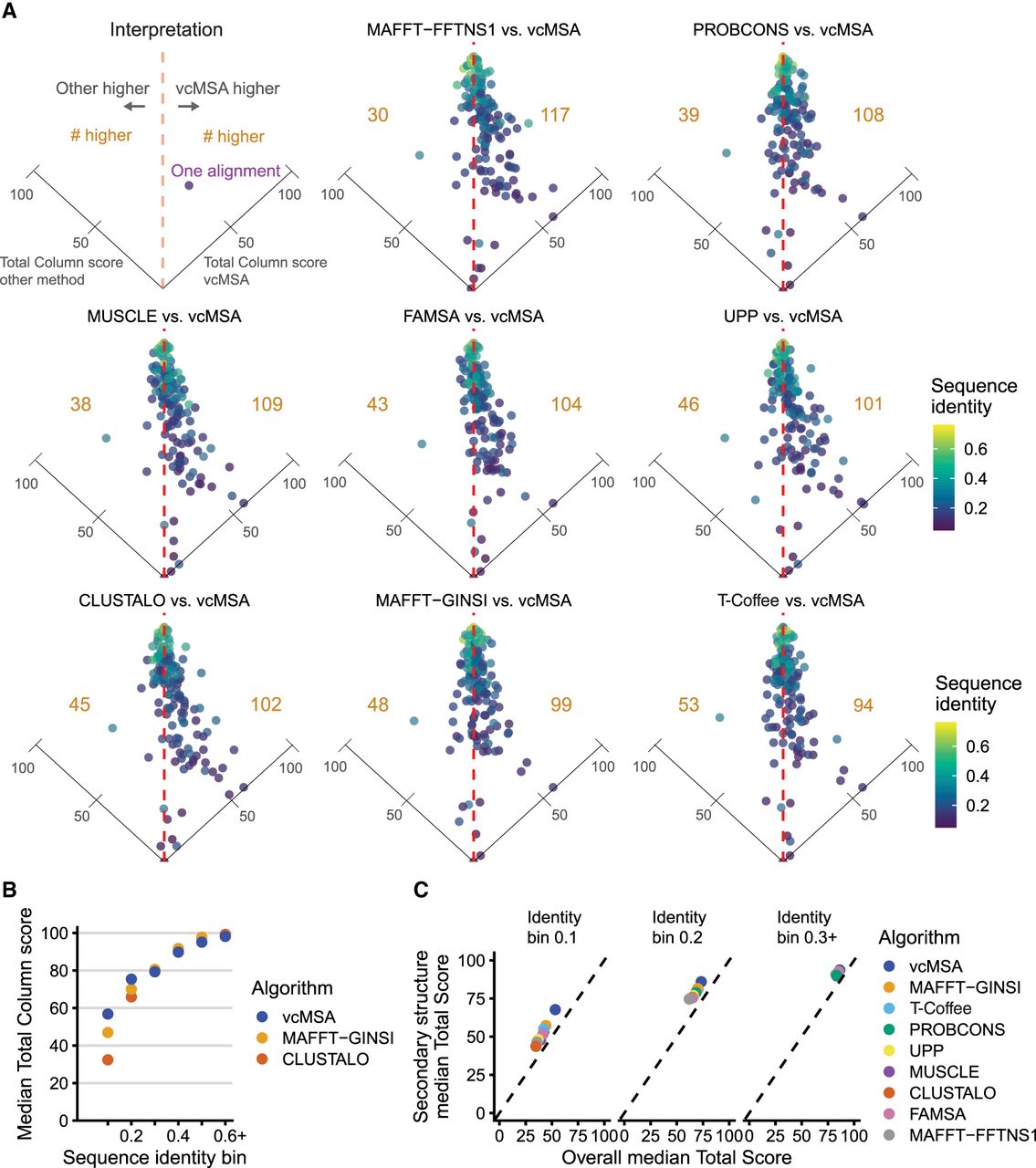
For low sequence identity alignments, we frequently exceed the alignment accuracy of existing algorithms. In our benchmarking, compared with other methods, we perform notably better on low sequence identity alignments. Figure 3B illustrates the performance of vcMSA against the two algorithms that score highest and lowest on sequences with identity < 0.2, MAFFT-GINSI and Clustal Omega, respectively. As sequence identity increases, vcMSA generally matches the accuracy of other algorithms. When we compute the total column score for structured portions of the gold-standard alignment compared with the full gold-standard alignment, it is apparent that vcMSA performs better on structured components (Fig. 3C). Further, vcMSA is specifically strong at structured regions of the lowest identity protein sets. This is likely because of the language model capturing structural properties of these low identity structured regions, even though their sequence identity is beyond the typical twilight zone limit.
Discussion
Although sequence aligners were some of the earliest bioinformatic algorithms, and although sequence alignment forms the basis of many computational biology analyses, MSA is not a solved problem. Current sequence alignment algorithms particularly struggle with alignments in the so-called twilight zone of sequence identity, in which structure and function is conserved, but amino acid sequence is not.
Here, we present vcMSA, a novel algorithm for MSA that diverges substantially from other approaches, and show that it is an improvement on the state of the art for some of the most challenging to align protein families. The ability to create accurate MSAs opens up these low sequence identity families to all the downstream bioinformatics applications that take alignments as input. Notably, the core vcMSA algorithm avoids standard features of the most widely used alignment algorithms, including gap penalties, substitution matrices, and guide trees. Although substitution matrices and gap penalties are standard components of alignment approaches, they are used to formalize optimization criteria and do not reflect fundamental properties of alignments. Instead, aligned amino acids within an MSA are meant to be those that have evolved from a shared ancestral amino acid. Protein language models may be especially suited to identify these analogous amino acids, as they capture structural and functional properties of amino acids via sequence context, much as natural language transformer models capture the semantics of words in a sentence based on other words (Vaswani et al. 2017). We find that vcMSA performs particularly well at aligning structured portions of sequences; it is possible that the less well-structured portions of sequences do not have specific functionally analogous positions in other sequences.
Protein alignment based on protein language representation appears to be most suited to global alignment for sets of protein sequences with similar structure and function, where the underlying meaning of the sequence is as consistent as possible. We have observed that alignment of a small subsequence of a protein (e.g., one domain) to a longer protein is often highly inaccurate, as the contextual embeddings for the small portion are sufficiently different from those computed for the longer protein. To produce the highest quality alignment, outlier sequences can be automatically detected based on sequence embeddings and removed from the alignment process. In these cases, we first use sequence representations to divide sequences into high similarity groups and detect outlier sequences. We then perform vcMSA on each cluster of sequences individually, producing high-quality subalignments. Although we currently use MAFFT-LINSI to combine subalignments produced by vcMSA, we are exploring methods to combine subalignments using purely language models. However, divergence in protein meaning between sequence groups complicates this process.
Additionally, for alignments in which start positions are not aligned, we find it helpful to add sequence padding to either end of the sequence before embedding, which reduces an observed first character effect, in which the first characters of each sequence tend to have the most similar embeddings, regardless of the sequence context of that first character (Supplemental Fig. 1A). In these cases, padding can substantially improve correctly aligned columns beyond the first column (Supplemental Fig. 1B). However, adding padding either substantially degrades or does not change alignment accuracy for protein sets in which the start positions align.
For certain protein groups, we observe a sequence-specific offset in amino acid embeddings (Supplemental Fig. 1C), even for sequences with relatively high sequence and structure similarity. On the other hand, we observe that fairly divergent protein sequences can result in embeddings without noticeable sequence-level effects. To reduce sequence specific effects, we consider the amino acids from each sequence as analogous to a batch and remove sequence-specific variation using batch-correction-style techniques (Supplemental Methods). Reducing sequence-specific effects with batch correction overall improves alignment quality (Supplemental Fig. 1D). Specifically, between pairs of sequences, batch correction improves the quality of RBHs, particularly for pairs of sequences with cosine similarity less than 0.95 (Supplemental Fig. 1E). Accordingly, by default, we use batch correction to remove sequence-specific offsets. In some cases, even with correction for sequence-specific offsets, alignment quality is poor. For example, batch correction improves the total column score of neuraminidases from 0.5 to 36.9. However, aligning neuraminidases from Influenza A and B separately and then combining subalignments allows a total column score of 64.7. Functional divergence in response to selective pressure in these two neuraminidases likely decreases the similarity of amino acid embeddings. Further research is needed to understand how to recognize and reduce sequence-specific effects when comparing amino acid embeddings across sequences.
Overall, we find that alignment quality and speed of construction is related to the quality of the RBHs between amino acid embeddings. To improve this network, we use MNCM to remove false-positive RBHs (Supplemental Fig. 2). However, it is likely that the RBH network could be improved in multiple ways, including through fine-tuning embeddings, use of other protein language models, and choice of embedding layers. On the other hand, because finding and filtering RBHs is largely effective for removing low similarity spurious matches, our approach is robust to changes in the similarity cutoffs for identifying amino acids to align. We find that alignments improve when merging neighboring columns in the alignment that contain amino acids from mutually exclusive sequences and whose amino acids have a minimal cosine similarity to each other.
As our implementation is a prototype, we have not substantially optimized our scripts for speed and memory usage, with much room for improvement in these areas. To identify similar amino acid positions at-scale between larger numbers of sequences, we can use tools developed for ultrafast nearest neighbor searches of millions and billions of vectors (Johnson et al. 2021). Additionally, our algorithm's runtime is greatly dependent on the number of iterations required to place all amino acids, which is related to the number of clustering errors (when an amino acid is sorted into a cluster that conflicts with the order of amino acids in the sequence). Currently, we use the very basic approach of finding connected components and choosing them to be clusters; we believe that slightly more sophisticated approaches (e.g., those requiring a higher edge density within clusters) would reduce errors in clustering and decrease runtimes.
Our prototype implementation of the vcMSA algorithm outperforms existing alignment algorithms at aligning a benchmark of sets of proteins. It is particularly successful at aligning groups of low similarity sequences, filling a gap in the capabilities of other algorithms. Another advantage of using amino acid embeddings is that we can additionally score the confidence of each column in the alignment by measuring the average cosine similarity of amino acid embeddings from the same column. This confidence score will allow filtering of alignments to only confidently aligned positions in downstream tasks such as phylogenetic tree building.
Here, we show both the utility of protein language embeddings in MSA and a new MSA algorithm fully orthogonal to existing approaches. We expect accuracy to only improve with optimal choice of amino acid representations and the development of larger language models that will more richly capture the identity of each amino acid. Overall, we anticipate that protein language models will play an important role in the next generation of methods for determining MSAs.
Software availability
Our modified version of the nf-benchmark (Garriga et al. 2019) is available at GitHub (https://github.com/clairemcwhite/nf-benchmark-vcmsa). A demonstration colab notebook is available at GitHub (https://github.com/clairemcwhite/vcmsa). Source code is distributed as a Python package vcmsa under an MIT free software license. We recommend obtaining vcmsa and associated files from the GitHub repository (https://github.com/clairemcwhite/vcmsa); however, it is also made available here as Supplemental Code. A demonstration Google colab notebook is additionally available at https://colab.research.google.com/drive/1h8WcsMzi8pYr-O3jW03knt7ePcv22o-F.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
C.D.M. acknowledges support from the Lewis-Sigler Institute for Integrative Genomics. M.S. acknowledges support from National Institutes of Health (NIH) R01-GM076275. We thank Joshua Akey for helpful discussions and also thank the Princeton University High Performance Computing Center for providing high-performance computing resources that have contributed to the research results reported in this paper.
Author contributions: Conceptualization and algorithmic design were by C.D.M. and M.S. Software implementation and result and figure generation were by C.D.M., with contributions from I.A.-G. C.D.M. and M.S. wrote the manuscript, with contributions from I.A.-G.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277675.123.
[3] Freely available online through the Genome Research Open Access option.
References
- ↵Altschul SF. 1991. Amino acid substitution matrices from an information theoretic perspective. J Mol Biol 219: 555–565. 10.1016/0022-2836(91)90193-A
- ↵Bepler T, Berger B. 2021. Learning the protein language: evolution, structure, and function. Cell Syst 12: 654–669.e3. 10.1016/j.cels.2021.05.017
- ↵Capra JA, Singh M. 2007. Predicting functionally important residues from sequence conservation. Bioinformatics 23: 1875–1882. 10.1093/bioinformatics/btm270
- ↵Capra JA, Singh M. 2008. Characterization and prediction of residues determining protein functional specificity. Bioinformatics 24: 1473–1480. 10.1093/bioinformatics/btn214
- ↵Chowdhury R, Bouatta N, Biswas S, Floristean C, Kharkar A, Roy K, Rochereau C, Ahdritz G, Zhang J, Church GM, 2022. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol 40: 1617–1623. 10.1038/s41587-022-01432-w
- ↵Csardi G, Nepusz T. 2006. The igraph software package for complex network research. Int J Complex Syst 1695: 1–9.
- ↵Dayhoff MO, Schwartz RM, Orcutt BC. 1978. A model of evolutionary change in proteins. Atlas Protein Seq Struct 5: 345–352.
- ↵Deorowicz S, Debudaj-Grabysz A, Gudyś A. 2016. FAMSA: fast and accurate multiple sequence alignment of huge protein families. Sci Rep 6: 33964. 10.1038/srep33964
- ↵Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res 15: 330–340. 10.1101/gr.2821705
- ↵Eades P, Lin X, Smyth WF. 1993. A fast and effective heuristic for the feedback arc set problem. Inf Process Lett 47: 319–323. 10.1016/0020-0190(93)90079-O
- ↵Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113. 10.1186/1471-2105-5-113
- ↵Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, Gibbs T, Feher T, Angerer C, Steinegger M, 2022. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans Pattern Anal Mach Intell 44: 7112–7127. 10.1109/TPAMI.2021.3095381
- ↵Felsenstein J. 2004. Inferring phylogenies, 2nd ed. Sinauer Associates, Sunderland, MA.
- ↵Feng D-F, Doolittle RF. 1987. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol 25: 351–360. 10.1007/BF02603120
- ↵Garriga E, Di Tommaso P, Magis C, Erb I, Mansouri L, Baltzis A, Laayouni H, Kondrashov F, Floden E, Notredame C. 2019. Large multiple sequence alignments with a root-to-leaf regressive method. Nat Biotechnol 37: 1466–1470. 10.1038/s41587-019-0333-6
- ↵Heinzinger M, Littmann M, Sillitoe I, Bordin N, Orengo C, Rost B. 2022. Contrastive learning on protein embeddings enlightens midnight zone. NAR Genom Bioinform 4: lqac043. 10.1093/nargab/lqac043
- ↵Henikoff S, Henikoff JG. 1992. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci 89: 10915–10919. 10.1073/pnas.89.22.10915
- ↵Johnson J, Douze M, Jégou H. 2021. Billion-scale similarity search with GPUs. IEEE Trans Big Data 7: 535–547. 10.1109/TBDATA.2019.2921572
- ↵Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589. 10.1038/s41586-021-03819-2
- ↵Kabsch W, Sander C. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22: 2577–2637. 10.1002/bip.360221211
- ↵Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. 10.1093/molbev/mst010
- ↵Katoh K, Toh H. 2007. PartTree: an algorithm to build an approximate tree from a large number of unaligned sequences. Bioinformatics 23: 372–374. 10.1093/bioinformatics/btl592
- ↵Littmann M, Heinzinger M, Dallago C, Weissenow K, Rost B. 2021. Protein embeddings and deep learning predict binding residues for various ligand classes. Sci Rep 11: 23916. 10.1038/s41598-021-03431-4
- ↵Liu K, Warnow TJ, Holder MT, Nelesen SM, Yu J, Stamatakis AP, Linder CR. 2012. SATé-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst Biol 61: 90. 10.1093/sysbio/syr095
- ↵Malucelli F, Ottmann T, Pretolani D. 1993. Efficient labelling algorithms for the maximum noncrossing matching problem. Discrete Appl Math 47: 175–179. 10.1016/0166-218X(93)90090-B
- ↵Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C. 2011. Protein 3D structure computed from evolutionary sequence variation. PLoS One 6: e28766. 10.1371/journal.pone.0028766
- ↵Marquet C, Heinzinger M, Olenyi T, Dallago C, Erckert K, Bernhofer M, Nechaev D, Rost B. 2022. Embeddings from protein language models predict conservation and variant effects. Hum Genet 141: 1629–1647. 10.1007/s00439-021-02411-y
- ↵Mirarab S, Nguyen N, Guo S, Wang L-S, Kim J, Warnow T. 2015. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J Comput Biol 22: 377–386. 10.1089/cmb.2014.0156
- ↵Miyata T, Yasunaga T. 1980. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J Mol Evol 16: 23–36. 10.1007/BF01732067
- ↵Morgenstern B, Prohaska SJ, Pöhler D, Stadler PF. 2006. Multiple sequence alignment with user-defined anchor points. Algorithms Mol Biol 1: 6. 10.1186/1748-7188-1-6
- ↵Morton JT, Strauss CEM, Blackwell R, Berenberg D, Gligorijevic V, Bonneau R. 2020. Protein structural alignments from sequence. bioRxiv 10.1101/2020.11.03.365932
- ↵Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443–453. 10.1016/0022-2836(70)90057-4
- ↵Nguyen ND, Mirarab S, Kumar K, Warnow T. 2015. Ultra-large alignments using phylogeny-aware profiles. Genome Biol 16: 124. 10.1186/s13059-015-0688-z
- ↵Notredame C, Holm L, Higgins DG. 1998. COFFEE: an objective function for multiple sequence alignments. Bioinformatics 14: 407–422. 10.1093/bioinformatics/14.5.407
- ↵Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217. 10.1006/jmbi.2000.4042
- ↵Nute M, Saleh E, Warnow T. 2019. Evaluating statistical multiple sequence alignment in comparison to other alignment methods on protein data sets. Syst Biol 68: 396–411. 10.1093/sysbio/syy068
- ↵Petti S, Bhattacharya N, Rao R, Dauparas J, Thomas N, Zhou J, Rush AM, Koo PK, Ovchinnikov S. 2023. End-to-end learning of multiple sequence alignments with differentiable Smith–Waterman. Bioinformatics 39: btac724. 10.1093/bioinformatics/btac724
- ↵Pitschi F, Devauchelle C, Corel E. 2010. Automatic detection of anchor points for multiple sequence alignment. BMC Bioinformatics 11: 445. 10.1186/1471-2105-11-445
- ↵Rao R, Bhattacharya N, Thomas N, Duan Y, Chen X, Canny J, Abbeel P, Song YS. 2019. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst 32: 9689–9701.
- ↵Rives A, Meier J, Sercu T, Goyal S, Lin Z, Liu J, Guo D, Ott M, Zitnick CL, Ma J, 2021. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci 118: e2016239118. 10.1073/pnas.2016239118
- ↵Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng 12: 85–94. 10.1093/protein/12.2.85
- ↵Rost B, Sander C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19: 55–72. 10.1002/prot.340190108
- ↵Sievers F, Higgins DG. 2020. QuanTest2: benchmarking multiple sequence alignments using secondary structure prediction. Bioinformatics 36: 90–95. 10.1093/bioinformatics/btz552
- ↵Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol 7: 539. 10.1038/msb.2011.75
- ↵Smith TF, Waterman MS. 1981. Identification of common molecular subsequences. J Mol Biol 147: 195–197. 10.1016/0022-2836(81)90087-5
- ↵Sonnhammer ELL, Eddy SR, Birney E, Bateman A, Durbin R. 1998. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26: 320–322. 10.1093/nar/26.1.320
- ↵Stärk H, Dallago C, Heinzinger M, Rost B. 2021. Light attention predicts protein location from the language of life. Bioinforma Adv 1: vbab035. 10.1093/bioadv/vbab035
- ↵Stebbings LA, Mizuguchi K. 2004. HOMSTRAD: recent developments of the Homologous Protein Structure Alignment Database. Nucleic Acids Res 32: D203–D207. 10.1093/nar/gkh027
- ↵Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, Kaiser Ł, Polosukhin I. 2017. Attention is all you need. Advances in neural information processing systems. In Proceedings of the 31st international conference on neural information processing systems (NIPS'17), pp. 6000–6010. Curran Associates Inc., Red Hook, NY. 10.5555/3295222.3295349.