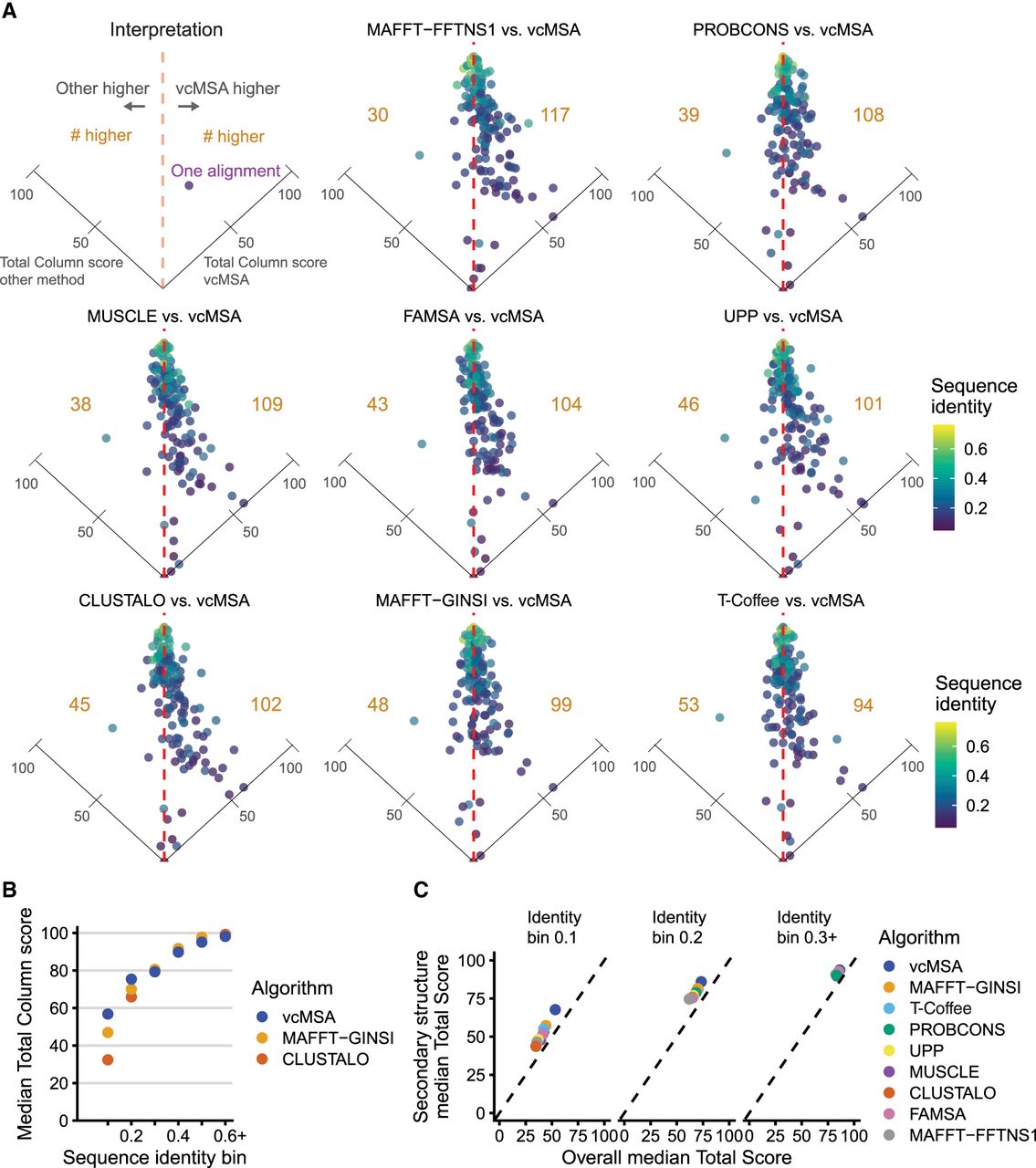
vcMSA alignments are frequently more accurate than previous methods, particularly for alignments with low levels of sequence identity. We benchmarked our implementation of vcMSA on sets of 20 sequences from each of 147 gold-standard multiple sequence alignments from the QuanTest2 data set. (A) We compare our method to eight state-of-the-art algorithms for sequence alignment. Each point corresponds to one protein MSA and is colored by its sequence identity. Each panel is a tilted scatterplot of relative performance of each other algorithm tested against vcMSA by total column score of produced alignments. Points to the right of the vertical dashed line are alignments where vcMSA outperformed the other method. The number of alignments to the left or right of the line is in gold. vcMSA performs better on more test sets than all other tested algorithms. The point lying on the right axis line is the Asp_Glu_race_D alignment, which all other algorithms perform poorly on (TC score ∼0). (B) We compare the median total column score of vcMSA alignments to MAFFT-GINSI and Clustal Omega alignments, grouping alignments by sequence identity bins. We observe increased scores for vcMSA at low sequence identities. (C) We show median total column scores for structured portions and full gold-standard reference alignments for all algorithms, separated by sequence identity bin.