
Abstract
Many cancer genomes are extensively rearranged with aberrant chromosomal karyotypes. Deriving these karyotypes from high-throughput DNA sequencing of bulk tumor samples is complicated because most tumors are a heterogeneous mixture of normal cells and subpopulations of cancer cells, or clones, that harbor distinct somatic mutations. We introduce a new algorithm, Reconstructing Cancer Karyotypes (RCK), to reconstruct haplotype-specific karyotypes of one or more rearranged cancer genomes from DNA sequencing data from a bulk tumor sample. RCK leverages evolutionary constraints on the somatic mutational process in cancer to reduce ambiguity in the deconvolution of admixed sequencing data into multiple haplotype-specific cancer karyotypes. RCK models mixtures containing an arbitrary number of derived genomes and allows the incorporation of information both from short-read and long-read DNA sequencing technologies. We compare RCK to existing approaches on 17 primary and metastatic prostate cancer samples. We find that RCK infers cancer karyotypes that better explain the DNA sequencing data and conform to a reasonable evolutionary model. RCK’s reconstructions of clone- and haplotype-specific karyotypes will aid further studies of the role of intra-tumor heterogeneity in cancer development and response to treatment. RCK is freely available as open source software.
The somatic mutations that drive cancer development range across all genomic scales, from single-nucleotide mutations through copy number aberrations and large-scale genome rearrangements (Stratton et al. 2009; Garraway and Lander 2013; Vogelstein et al. 2013; Raphael et al. 2014). Whole-genome sequencing of tumor samples has enabled the detection of all classes of somatic mutations; however, specialized algorithms are required to identify each class of mutations from the short DNA sequence reads obtained by current technologies. In addition, nearly all cancer sequencing to date has been of bulk tumor tissue, which is generally a mixture of normal (noncancerous) cells and (sub)populations of cancerous cells, or clones, that often are not genetically identical. Quantifying this intra-tumor heterogeneity is essential for understanding the processes that drive cancer development and also helps inform treatment strategies (Aparicio and Caldas 2013; McGranahan and Swanton 2015; Patch et al. 2015).
Here, we consider the problem of describing the large-scale organization of one or more cancer genomes that are derived from a normal human reference genome via copy number aberrations and rearrangements. The large-scale organization of a cancer genome is described by two features. First is the number of copies of each segment of the genome. Many methods (e.g., Van Loo et al. 2010; Boeva et al. 2012; Carter et al. 2012; Nik-Zainal et al. 2012; Fischer et al. 2014; Ha et al. 2014; Oesper et al. 2014; Zaccaria and Raphael 2020) have been developed to identify copy number values for heterogeneous, bulk tumor samples. Second is genome rearrangements (e.g., chromosomal inversions and translocations) that link together distant segments of the normal genome. Many methods have been developed to predict the novel adjacencies resulting from such rearrangements (e.g., Chen et al. 2009; Quinlan et al. 2010; Wang et al. 2011; Rausch et al. 2012; Sindi et al. 2012; English et al. 2014; Layer et al. 2014; Ritz et al. 2014; Zheng et al. 2016; Huddleston et al. 2017; Spies et al. 2017; Elyanow et al. 2018; Nattestad et al. 2018; Sedlazeck et al. 2018; Wala et al. 2018). However, these methods do not distinguish between adjacencies from different homologous chromosomes or from different cancer clones within a bulk sample, that is, they assume that the human genome is haploid and that the tumor is homogeneous.
A more challenging problem is to integrate and reconcile the information about segment copy numbers and novel adjacencies into genome karyotypes, or the alignment of cancer genome and the healthy genome that depicts the number of occurrences of every segment in the cancer genome, and the adjacencies between these segments on the cancer genome. Multiple methods have been developed to reconstruct cancer genome karyotypes, including PREGO (Oesper et al. 2012), Weaver (Li et al. 2016; Rajaraman and Ma 2018), ReMixT (McPherson et al. 2017), Karyotype Reconstruction (Eitan and Shamir 2017), SVclone (Cmero et al. 2020), and the method of Eaton et al. (2018). However, each of these methods relies on simplifying assumptions that do not adequately address the challenges in real cancer sequencing data. For example, SVclone (Cmero et al. 2020) focuses solely on inferring genome-specific copy numbers for novel adjacencies, without attempting to reconstruct complete karyotypes of the derived genomes. PREGO (Oesper et al. 2012) and Karyotype Reconstruction (Eitan and Shamir 2017) assume that the human reference genome is haploid, thus losing important information about alleles involved in rearrangements. Weaver (Li et al. 2016; Rajaraman and Ma 2018) assumes that the cancer sample contains only a single derived genome (with a possible admixture of the reference genome) and lacks a proper support of reciprocal novel adjacencies, which can emerge both from copy neutral somatic rearrangements (e.g., inversions, balanced translocations, and so forth), as well as from more complex “catastrophic rearrangements” such as chromoplexy and chromothripsis (Berger et al. 2011; Stephens et al. 2011; Baca et al. 2013; Hirsch et al. 2013; Weinreb et al. 2014; Oesper et al. 2018). ReMixT (McPherson et al. 2017) allows for tumor heterogeneity, but fixes the number of derived genomes in the observed cancer sample to 2. Moreover, although ReMixT aims to infer genome- and allele-specific segment copy numbers for a 2-genome sample (with a possible admixture of the reference genome), the genome-specific copy numbers for novel adjacencies that are inferred by ReMixT lack information about which homologous copies of the segments are actually involved in observed novel adjacencies. Last, Weaver and ReMixT produce karyotypes with biologically unlikely scenarios in which rearrangements occur repeatedly at the same homologous loci in different cancer clones. We summarize these limitations of existing methods in Supplemental Table S1.
Here, we propose a novel algorithm, Reconstructing Cancer Karyotypes (RCK), for deriving the karyotypes of cancer genomes in a heterogeneous tumor sample from second-generation (and third-generation, when available) sequencing data. RCK distinguishes itself from existing methods by several features, including (1) support for a diploid reference genome that distinguishes between alleles of segment copy numbers and novel adjacencies; (2) joint inference of both segment and adjacency copy numbers in both a clone- and haplotype-specific manner; (3) comprehensive support for sample heterogeneity ranging from homogeneous samples with a single derived genome to heterogeneous samples with an arbitrary number of clones; (4) a somatic evolutionary model based on a generalization of the infinite sites assumption; and (5) ability to incorporate groups of novel adjacencies from third-generation sequencing technologies into the inference model. We show the advantages of RCK on a data set of 17 primary and metastatic prostate cancer samples. We show that RCK infers more plausible karyotypes than ReMixT, and that the RCK inferred karyotypes have allele-specific segment copy numbers that agree with leading copy number inference algorithms.
Results
RCK algorithm
We introduce Reconstructing Cancer Karyotypes (RCK), an algorithm to construct the large-scale organization of one or more cancer genomes present in a bulk tumor sample. We assume that each cancer genome in the sample arises from a sequence of somatic genome rearrangements and copy number aberrations that transform a healthy normal genome into a cancer genome. As a result of these somatic mutations, each cancer genome can be represented as a karyotype graph, or more briefly, a karyotype. A karyotype graph includes (1) a collection of contiguous segments from the human reference genome, each segment with a label (A or B) distinguishing the two homologous chromosomes; (2) an integer copy number for each segment; (3) a collection of adjacencies that join the ends of segments; and (4) an integer copy number for each adjacency. The karyotype graph describes an alignment between the cancer genome and healthy genome (analogous to the breakpoint graph in genome rearrangement studies) (Alekseyev and Pevzner 2009; Avdeyev et al. 2016). The karyotype graph also represents the information about the cancer genome sequence that can be inferred from DNA sequencing technologies whose read lengths are shorter than the length of genome rearrangements.
RCK solves the following Cancer Karyotype Reconstruction Problem: given allele-specific segment copy numbers and a list of novel adjacencies (i.e., pairs of genomic loci that are measured as adjacent in the cancer genome but distant in the normal reference) from a bulk tumor sample, derive karyotype graph(s) for the cancer genome(s) present in the tumor sample. Two major challenges must be addressed in developing an algorithm to solve this problem. The first challenge is that methods for inferring allele-specific copy numbers from bulk tumor sequencing data do not preserve the allelic information across multiple adjacent segments. Specifically, these methods output a pair of copy number vectors, and , where the pair of integers indicates the number of copies of each of the two homologous copies of segment j from the reference genome that are present in the cancer genome. However, each of these pairs is unordered: for each segment j, it is unknown whether is the number of copies from the maternal chromosome or the paternal chromosome; moreover, the assignment of to either the maternal or paternal chromosome is independent for each j. The second challenge is that the many methods for inferring novel adjacencies from bulk tumor sequencing data generally do not include two important attributes in their output: (1) the alleles (maternal or paternal) that are joined by the adjacency, and (2) the copy number(s) of the adjacency in each genome in the sample. Because of this incomplete information in the allele-specific copy numbers and novel adjacencies, cancer genome karyotypes are not directly available.
RCK derives optimal cancer genome karyotype(s) from allele-specific copy numbers and novel adjacencies by solving an optimization problem on a graph, called the Diploid Interval Adjacency Graph (DIAG) (Fig. 1). The vertices of the DIAG are extremities, or the positions in the human reference genome of the endpoints of the segments that are rearranged to form the cancer genomes present in the sample. Specifically, if we enumerate the segments of the reference genome , then each segment j has the form , where and are extremities. The label t indicates that the extremity is the tail, or starting coordinate of the segment in the reference genome, whereas the label h indicates the head, or ending coordinate in the reference genome. A haplotype label H ∈ {A, B} indicates which copy of the two homologous chromosomes in the reference (A or B) is the source of the segment. Adjacent extremities of consecutive segments that follow each other along the chromosome in the genome constitute an adjacency. We distinguish between two types of adjacencies: reference adjacencies that are present in the reference genome, and novel adjacencies that are not present in the reference genome. Thus, the DIAG has three types of edges: (1) segment edges join extremities from a segment; (2) reference adjacency edges join extremities of adjacent segments on the reference genome; and (3) novel adjacency edges join extremities that are not adjacent in the reference genome, where H, H′ ∈ {A, B} and σ, σ′ ∈ {t, h}. Importantly, a measured novel adjacency is generally unlabeled, having the form and lacking allelic information. To model this uncertainty, we add all four possible labeled versions of the adjacency (, , , and ) to the DIAG. Supplemental Table S2 summarizes the notation used to describe the DIAG.
Overview of the RCK algorithm. The inputs to RCK (white dotted boxes) are clone- and allele-specific copy numbers (top left) and novel adjacencies (top right) from bulk tumor samples that are derived from alignments of DNA sequencing (top) reads using existing tools. The RCK algorithm (blue shaded elements) builds a diploid interval adjacency graph integrating copy number and novel adjacency information (for details, see Methods). RCK then solves a mixed-integer linear program (MILP) to find an optimal assignment of segment copy numbers and novel adjacencies to alleles and clones, subject to copy number balance on segment ends and satisfying evolutionary constraints from a generalized infinite sites model. Constraints on groups of novel adjacencies from the third-generation sequencing technologies may optionally be included. The outputs of RCK are clone- and haplotype-specific cancer genome karyotypes.
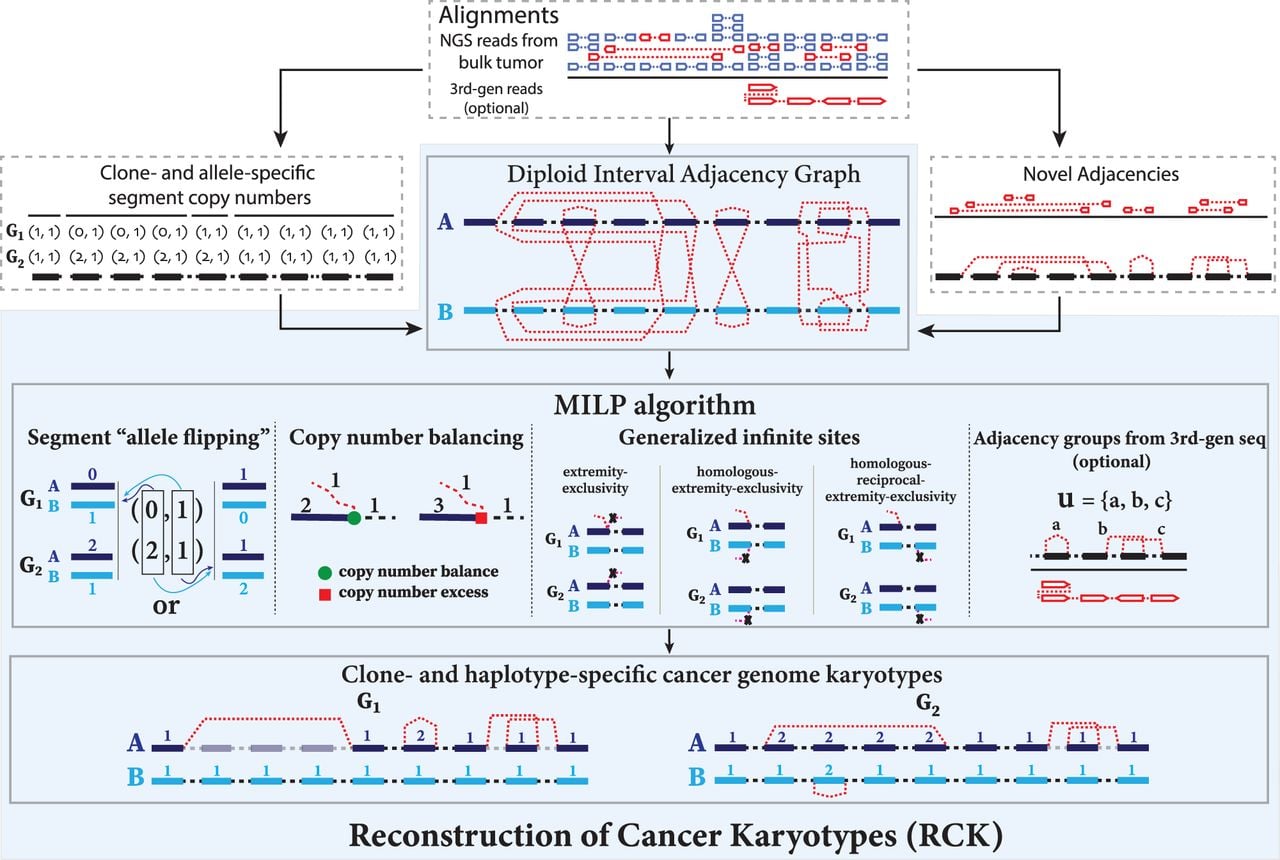
A chromosome in the cancer genome corresponds to a walk in the DIAG that alternates between segment edges and reference/novel adjacency edges, and where the number of times every segment/adjacency edge is visited encodes the respective segment/adjacency copy number (see Methods, “Diploid interval adjacency graph”). Thus, all vertices (except telomere vertices) should satisfy the copy number balance condition: the copy number of the incident segment edge equals the sum of the copy numbers of the incident reference edge and novel adjacency edge(s).
RCK solves the Cancer Karyotype Reconstruction Problem of finding an edge multiplicity μG (e) for each edge e and each cancer genome G such that (1) each extremity (vertex ν) satisfies the copy number balancing conditions (Equations [5] and [6] in Methods); (2) the copy numbers μG (jA) and μG (jB) of homologous segments jA and jB are approximately equal to the allele-specific copy numbers ( and ); and (3) most of the novel adjacencies are present in at least one genome, that is, μG (e) ≥ 0 for novel adjacency edge e in at least one genome G. However, there are often numerous solutions to this problem owing to the lack of A/B labels on the measured novel adjacencies with each measured novel adjacency generating four edges in the DIAG. Selecting one of these four possible allele-specific novel adjacencies independently for each measured novel adjacency often leads to biologically implausible solutions.
To address this ambiguity, RCK imposes several constraints on the inferred karyotypes that are motivated by the somatic evolutionary process of cancer. In particular, RCK uses conditions on allowed novel adjacencies that are derived from a generalization of the infinite sites (IS) assumption commonly used in evolutionary studies. The infinite sites assumption is that a mutation does not occur at the same locus more than once during the course of evolution. The locus of a large-scale genome rearrangement is not apparent and could be defined as either (or both) of the genomic positions of the extremities in the adjacency as well as adjacent genomic positions of “reciprocal” extremities. We define multiple constraints on the extremities that may be involved in novel adjacencies (Fig. 1), which generalize the infinite sites assumption to the case of multiple genomes that are derived from a diploid reference genome by a sequence of large-scale genome rearrangements. First, extremity-exclusivity is the constraint that an extremity is involved in at most one novel adjacency. Second, homologous-extremity-exclusivity is the constraint that an extremity and its homolog cannot both be involved in a novel adjacency. Third, homologous-reciprocal-extremity-exclusivity is the constraint that an extremity and its reciprocal mate of the homologous chromosome cannot both be involved in a novel adjacency. Methods that rely on weaker forms of the infinite sites assumption can yield implausible genome reconstructions, as we will show below.
RCK uses a mixed-integer linear program (see Supplemental Methods, “MILP formulation”) to find edge multiplicities μG (e) satisfying conditions (1), (2), and (3) above while also requiring that the novel adjacencies inferred to be present (μG (e) > 0) satisfy the generalized infinite sites constraints. RCK also allows for grouping of novel adjacencies that are measured to be present on the same cell or long read when such information is available from third-generation sequencing technologies, for example, single-cell sequencing, linked read sequencing (Zheng et al. 2016; Spies et al. 2017; Elyanow et al. 2018), or long-read sequencing (English et al. 2014; Ritz et al. 2014; Huddleston et al. 2017; Sedlazeck et al. 2018). See Methods, “Third-generation sequencing technologies and novel adjacency groups” for further details.
Evaluation of RCK on simulated data
We first evaluate RCK on simulated cancer genomes. We simulated bulk tumor samples containing up to two rearranged cancer genomes, or clones. The simulation starts with a normal diploid reference genome and a somatic phylogenetic tree as an input, and then sequentially applies random genome rearrangements along the branches of the tree. The simulated genome rearrangements (Supplemental Fig. S1A) include simple rearrangements (e.g., deletion, duplication, inversion, translocation, whole chromosome amplification/loss (WCA/L)) and complex rearrangements (e.g., breakage-fusion-bridge [BFB], whole-genome duplication [WGD], chromothripsis, and chromoplexy). Following these rearrangements, the derived genomes of each clone are recorded on the leaves of the tree (Supplemental Fig. S1B). Every simulated tumor sample includes at least one complex rearrangement that occurred early and was shared by all derived cancer genomes in the sample, consistent with reports on the early occurrence of “catastrophic” rearrangements in cancer genomes (Cortés-Ciriano et al. 2020; Gerstung et al. 2020). Although the simulator is capable of explicitly enforcing the generalized infinite sites constraints described above, we found that randomly simulated rearrangements satisfied these constraints. Further details of simulations are in Methods, “Simulating rearranged cancer samples.”
We evaluated RCK’s performance on two different types of simulated data. In the first, there are no errors in the novel adjacencies and allele-specific segment copy numbers that are input to RCK. However, the novel adjacencies are missing information about the clone(s) containing the adjacency as well as the haplotype involved in the adjacency. Similarly, the segment copy numbers are missing the haplotype information. We say that this input data has clone and haplotype information loss (CHIL) (Supplemental Fig. S1C). On this data we find that RCK outputs karyotypes that use all input novel adjacencies and whose haplotype-labeled copy numbers agree with the simulated copy numbers, for both adjacencies and segments.
Next, we simulated data with errors in novel adjacencies and segment copy numbers. Specifically, we introduce uncertainty into coordinates of novel adjacency and include 10% spurious adjacencies, resulting in a set of novel adjacencies. We introduce errors in copy number profiles by averaging the values in true segment copy number profile C over 50 kbp fragments, and also perturbing 5% of fragment copy number values by ±1, resulting in a copy number profile (Supplemental Fig. S1C).
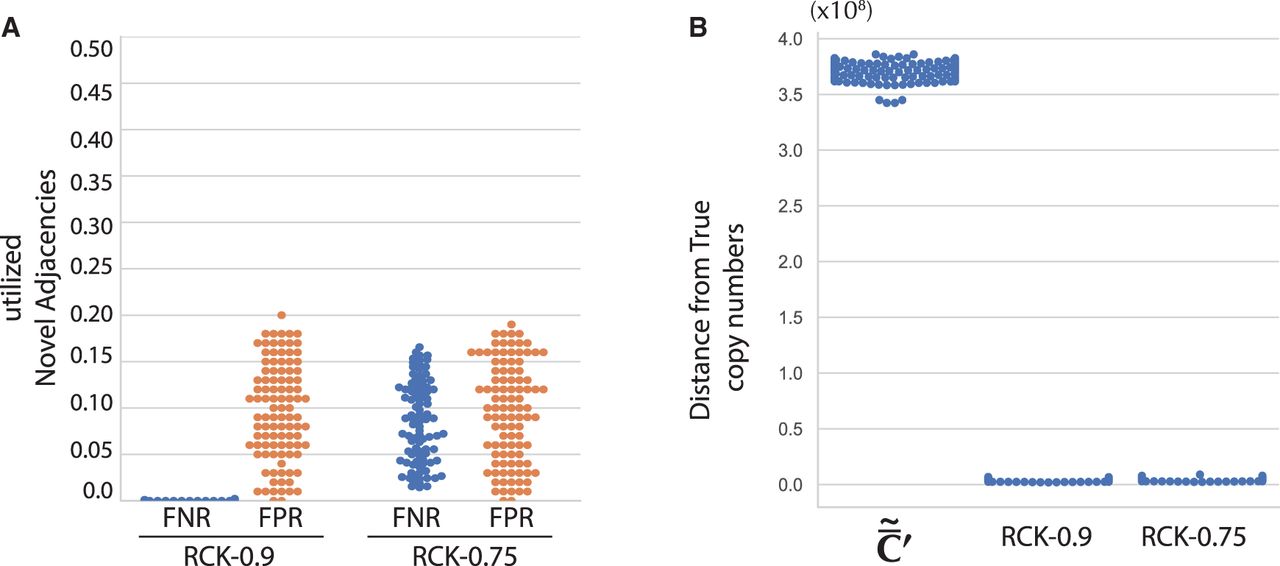
We ran RCK with input adjacency utilization parameter P = 0.9 and P = 0.75, that is, at least a fraction P of the input novel adjacencies set must be present in at least one of the derived genomes in a sample. We then compared inferred copy numbers for novel adjacencies and segments in a haplotype-specific manner (up to haplotype symmetries; see Methods, “Sequencing of rearranged cancer genomes”) with the true values in the input. We found that karyotypes reconstructed by RCK (P = 0.9) use almost all true novel adjacencies with an average false negative rate (FNR) of < 0.003, and rarely incorporate spurious input novel adjacencies with average false positive rate (FPR) of < 0.12 (Fig. 2A). When P = 0.75 (less than the actual fraction 0.9 of true novel adjacencies in the input set ), the karyotypes reconstructed by RCK do not use a small fraction of true novel adjacencies with an average FNR of <0.1, and the FPR remains low with an average of < 0.11 (Fig. 2; Supplemental Fig. S2A). We also found that the segment copy numbers output by RCK are closer to the true values using both a length-weighted segment copy number distance (Equation [11] in Methods; Fig. 2B), and a comparison of copy number states (i.e., amplification, neutral, and loss) (Supplemental Fig. S2B,C).
Results of RCK on simulated bulk tumor samples with two clones. (A) False negative rate (FNR) and false positive rate (FPR) of novel adjacencies used by RCK using adjacency utilization parameter P = 0.9 (RCK-0.9) and P = 0.75 (RCK-0.75). (B) Length-weighted segment copy number distances between input copy numbers () and karyotypes inferred by RCK.
We also observe that when the only source of error in the input segment copy numbers is a result of fragment-size averaging, both the FPR and FNR of novel adjacencies remain low (Supplemental Fig. S3A), and segment copy numbers inferred by RCK are closer to the true values in the simulated cancer samples (Supplemental Fig. S3B,C). Last, because of the generalized IS constraints, RCK correctly assigns groups of reciprocal novel adjacencies, generated by single chromothripsis k-break event, to the same haplotype-of-origin (e.g., Supplemental Fig. S4).
Evaluation of RCK on prostate cancer
We analyze a cancer sequencing data set from Gundem et al. (2015), which consists of whole-genome sequencing data from 49 samples from 10 metastatic prostate cancer patients. Segment copy numbers inferred by Battenberg (Nik-Zainal et al. 2012) and novel adjacencies were obtained from Gundem et al. (2015). We also applied HATCHet (Zaccaria and Raphael 2018) to infer allele-specific copy numbers by joint analysis across all sequenced samples from the same patient. We analyzed the 17 samples for which both Battenberg and HATCHet agreed on the number of clones present. We aligned the positions of extremities of segments from Battenberg or HATCHet to the positions of extremities from novel adjacencies. See “Deriving extremities and novel adjacencies from data” in Methods for further details. We divided the cancer samples into two groups according to the number of tumor clones predicted by both Battenberg and HATCHet: homogeneous samples containing only one tumor clone (samples A21g, A21h, A24c, A24d, A24e, A34a, A34d); and heterogeneous samples containing two tumor clones (samples A10c, A12c, A12d, A17d, A31a, A31d, A31e, A31f, A32e, A34c). Notably, there was only one sample (A12c) for which Battenberg and HATCHet disagreed on the presence of a WGD.
We compare RCK to ReMixT (McPherson et al. 2017), an existing method that both derives multiple tumor clones from bulk sequencing data and distinguishes between homologous chromosomes. ReMixT infers clone- and allele-specific copy numbers for segments, as well as clone-specific copy numbers for novel adjacencies. Importantly, ReMixT does not infer haplotype A/B labels for the extremities that are involved in each novel adjacency. We will show below that this lack of assignment of each novel adjacency to a homologous chromosome leads to unusual genome reconstructions in many cases.
For each sample, we ran RCK requiring that (1) the only telomeres in the inferred cancer genomes are telomeres from the reference genome (i.e., extremities that are not the endpoints of reference chromosomes have copy number balance); and (2) at least a fraction P of the input novel adjacencies are present in at least one of the derived genomes in a sample, for P = 1.0, 0.9, 0.75, 0.5. ReMixT does not allow control over telomeres or the fraction of novel adjacencies; thus, we ran ReMixT using default parameters.
Heterogeneous tumor samples
We compared the karyotypes inferred by RCK and ReMixT on the 10 samples from the heterogeneous group. First, we compared the segment copy numbers inferred by RCK and ReMixT to the allele-specific copy numbers from HATCHet and Battenberg, using a length-weighted segment copy number distance (Equation [11] in Methods). We found that in all but three cases (samples A10c, A12c, and A17d with RCK parameter P = 1.0), the segment copy numbers inferred by RCK are closer to the copy numbers from HATCHet (Fig. 3A) or Battenberg (Supplemental Fig. S5A), compared to the segment copy numbers inferred by ReMixT. In four samples (A31a, A31d, A31e, and A31f), copy numbers inferred by ReMixT have an extremely large distance to HATCHet (or Battenberg) copy numbers. Both HATCHet and Battenberg inferred a whole-genome duplication (WGD) in these four samples. Although ReMixT also infers high copy number values and many copy number changes in these four samples, the large copy number distances indicate the ReMixT’s inferred copy numbers do not seem to align well with copy numbers expected from a WGD. We also observe a large and consistent decrease in copy number distance when we require RCK to use all novel adjacencies (P = 1) versus when we allow a small fraction of novel adjacencies to be excluded (P = 0.9). The distance is largely stable for P < 0.9, showing that RCK is not overfitting the observed copy number values by excluding high fractions of adjacencies. Finally, note that the total length of segments for which RCK (P = 0.9) changed the copy numbers input by HATCHet or Battenberg is on average less than the overall inferred length-weighted segment copy number distances; this is because RCK changes the copy number of some segments by more than 1 (Supplemental Tables S3, S4).
Comparison of RCK and ReMixT on heterogeneous prostate cancer samples. (A) Length-weighted segment copy number distances between segment copy numbers from HATCHet and segment copy numbers output by ReMixT and RCK. (B) Fractions of novel adjacencies from input that are inferred to be present by ReMixT or RCK for each sample in the heterogeneous group. RCK used segment copy numbers from HATCHet in input and novel adjacency utilization parameter P = 1.0, 0.9, 0.75, 0.5.
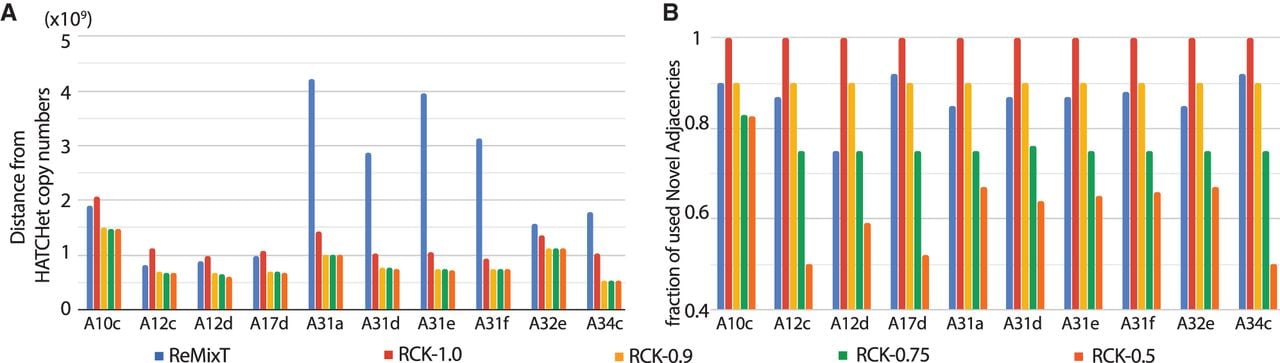
Next, we compared the fraction of input novel adjacencies that were contained in the karyotypes constructed by ReMixT and RCK. This fraction ranged from 0.75 to 0.92 for ReMixT (Fig. 3B) compared to a range from 0.5 to 1.0 for RCK, with the lower bound for RCK explicitly controlled via the P parameter. We observe that RCK frequently uses more novel adjacencies than the minimum required (value of P). This occurs on 7/10 cancer samples (A10c, A12d, A31a, A31d, A31e, A31f, A32e) with HATCHet copy numbers in input and P = 0.75 or P = 0.5, and 6/10 samples with Battenberg copy numbers in input. RCK’s incorporation of novel adjacencies at a higher proportion than the minimum required fraction P suggests that RCK is selectively including those novel adjacencies required to achieve copy number balance.
Next, we analyzed the number of novel (i.e., nonreference) telomeres in the karyotypes inferred by ReMixT. We observed that the karyotypes inferred by ReMixT have a substantially large numbers of inferred nonreference telomeres (ranging from 41 to 133 per genome) (Supplemental Fig. S6). In contrast, RCK required derived chromosomes to start and end at telomeres of the reference genome; thus, RCK karyotypes have no novel telomeres. Karyotypes reconstructed by ReMixT correspond to highly unlikely cancer genomes having dozens or even hundreds of linear chromosomes with novel telomeres. This large number of novel telomeres contradicts the recent PCAWG study (Sieverling et al. 2020) of more than 2500 cancer genomes, which reported that novel telomeres in prostate cancer were rare.
Finally, we examined the number of violations of the generalized IS constraints in the karyotypes inferred by RCK and ReMixT. By construction, RCK karyotypes have no such violations. In contrast, we identified numerous violations of generalized IS conditions in the ReMixT karyotypes, which we categorize into three types. The first type of violation is an intra-genome violation of the homologous-extremity-exclusivity constraint. This violation occurs when the inferred segment copy numbers require that a novel adjacency a be assigned both a label A and a label B to achieve copy number balance (Fig. 4A). This situation requires that at least two large-scale somatic rearrangements occurred independently at the same genomic position on both homologous chromosomes, which is highly unlikely. We find that karyotypes reconstructed by ReMixT contain such violations in 6/10 samples, ranging from 1 to 8 violations per genome, and from 1 to 12 violations per sample (Fig. 4B).
ReMixT karyotypes from heterogeneous prostate cancer samples have numerous violations of the generalized infinite sites constraints. In A, C, and E, solid edges represent segment edges, black-dashed edges represent reference adjacency edges, and red dashed edges represent novel adjacency edges. Integer values indicate copy numbers of corresponding segment and adjacency edges. (A) An intra-genome violation of the homologous-extremity-exclusivity constraint. To achieve copy number balance, both homologous vertices and from genome Gi must be involved in novel adjacencies. (B) Number of novel adjacencies that violate the intra-genome homologous-extremity-exclusivity constraint in each cancer karyotype inferred by ReMixT in each sample. (C) An inter-genome violation of the homologous-extremity-exclusivity constraint. To achieve copy number balance, both homologous vertices and (in different genomes) must be involved in novel adjacencies. (D) The fraction x/y, where x is the number of novel adjacencies that violate the inter-genome homologous-extremity-exclusivity constraint (on at least one of the extremities involved in a novel adjacency) in ReMixT karyotypes, and y is the total number of novel adjacencies reported by ReMixT as being present in both genomes. (E) A violation of the intra-genome homologous-reciprocal-extremity-exclusivity constraint. To achieve copy number balance, both homologous-reciprocal vertices and must be involved in novel adjacencies. Inter-genome violations of the homologous-reciprocal-extremity-exclusivity constraint are also possible (Supplemental Fig. S17). (F) Fraction x/y, where x is the number of reciprocal locations with violations of either intra- or inter-genome (or both) homologous-reciprocal-extremity-exclusivity constraint in ReMixT karyotypes; and y is the total number of reciprocal locations that both have novel adjacencies in ReMixT karyotypes.
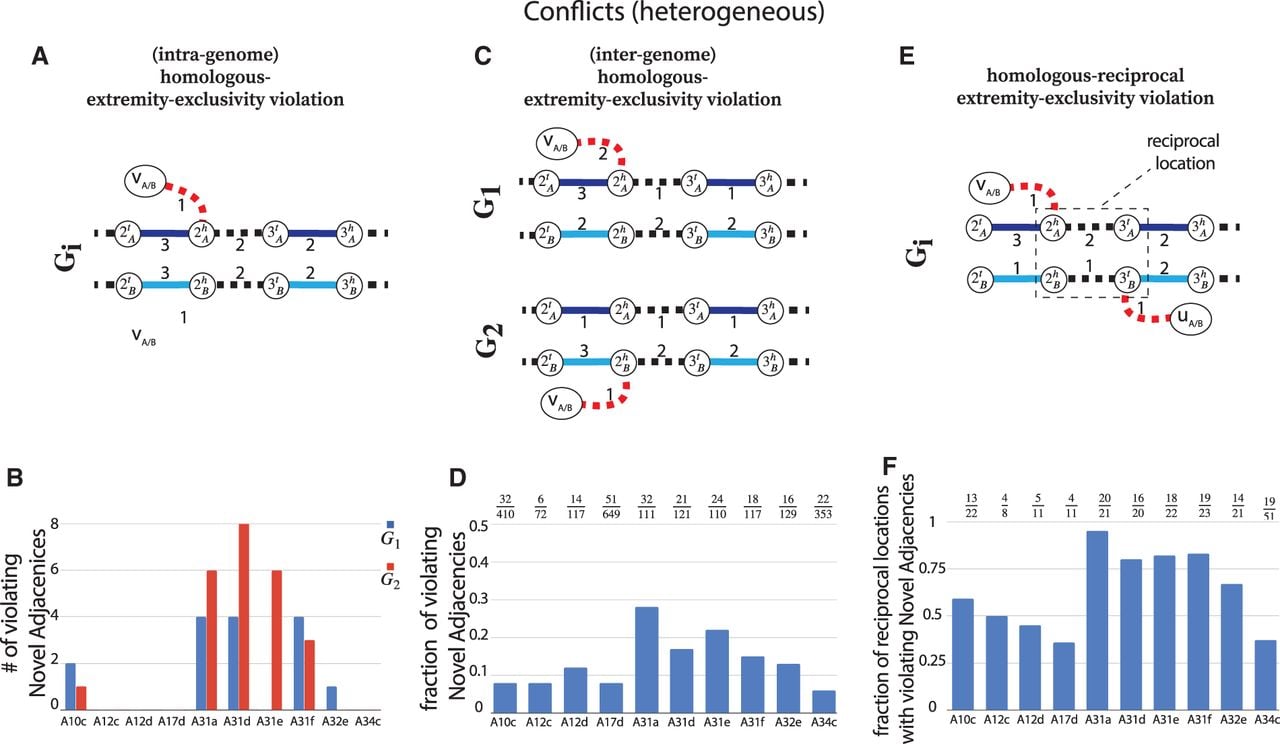
The second type of violation is an inter-genome violation of the homologous-extremity-exclusivity constraint (Fig. 4C). This violation occurs when a novel adjacency a is reported as being present in more than one genome in the sample, but a label A must be assigned to at least one a’s extremities in one genome, and a label B must be assigned to at least one a’s extremities in another genome. This situation requires that at least two large-scale somatic rearrangements occurred independently at the same homologous genomic location in two different tumor clones, which is highly unlikely. We found that the karyotypes produced by ReMixT had such violations in all samples, with a substantial fraction (ranging from 0.06 to 0.28) of novel adjacencies containing such violations (Fig. 4D).
The third type of violation concerns pairs of reciprocal novel adjacencies. For a pair a = {x, jh}, b = {(j + 1)t, y} of reciprocal novel adjacencies that involve adjacent extremities jh and (j + 1)t on the reference genome, possible violations of generalized IS include intra/inter-genome violation of the homologous-extremity-exclusivity or intra/inter-genome violation of the homologous-reciprocal-extremity-exclusivity constraints (Fig. 4E), or both. Any such violation requires that at least two large-scale somatic rearrangements occurred independently on the same or homologous genomic location both producing pairs of reciprocal novel adjacencies, a situation which is highly unlikely. We found that karyotypes produced by ReMixT had such violations in all samples; furthermore, in 6/10 samples, more than half of reciprocal novel adjacencies had such violations (Fig. 4F).
Homogeneous tumor samples
We compared the karyotypes inferred by RCK and ReMixT on the seven prostate cancer samples from the homogeneous group and analyzed the karyotypes output by both methods, following the procedures described above for the heterogeneous samples. Because ReMixT assumes that an input sample contains exactly two cancer clones, ReMixT’s results disagree with both Battenberg’s and HATCHet’s predictions of one cancer clone in these samples. Thus, we compared the segment copy number profiles of the clone inferred by ReMixT with the highest cellular prevalence in each sample with the copy number profiles inferred by Battenberg and HATCHet. We found that on every sample in the homogeneous group, the segment copy numbers inferred by RCK (with P ≤ 0.9) are more similar to the copy numbers from Battenberg (Supplemental Fig. S7A) and HATCHet (Supplemental Fig. S7B) compared to the segment copy numbers inferred by ReMixT. The fraction of input novel adjacencies that were present in the karyotypes inferred by ReMixT ranged from 0.82 to 0.94, compared to a range of 0.5 to 1.0 for RCK (Supplemental Fig. S8). As in the heterogeneous samples, we observed that segment copy number distances are largest for RCK when we require RCK to use all novel adjacencies (P = 1, a larger proportion than used in ReMixT), but that the distances decrease and stabilize when some novel adjacencies are excluded (P ≤ 0.9).
Similar to the heterogeneous samples, we also observed that karyotypes inferred by ReMixT had implausible features including a large number (and multiplicity) of novel telomeres (Supplemental Fig. S9) and violations of the generalized infinite sites constraints (Supplemental Fig. S10). In contrast, karyotypes inferred by RCK had no such issues. Overall, our analysis of inferred cancer genomes karyotypes in the homogeneous group aligned with the findings for the heterogeneous group.
Complex genome rearrangements in prostate cancer
We looked for evidence of complex rearrangements that involve simultaneous double-stranded DNA breakages at three or more genomic locations in the prostate cancer samples. Such complex rearrangements—including insertional duplications, chromoplexy, and chromothripsis—have recently been reported in multiple cancer types, including prostate cancer (Stephens et al. 2011; Baca et al. 2013; Hirsch et al. 2013; Weinreb et al. 2014; Oesper et al. 2018) and can affect genes and other functional genetic elements with important roles in cancer development and prognosis (Shen 2013; Fontana et al. 2018). Complex rearrangements are not directly observed in short-read DNA sequencing data, but rather must be inferred from the pattern of novel adjacencies that are created during such rearrangements. Specifically, under the infinite sites assumption, any pair a = {x, jh}, b = {(j + 1)t, y} of reciprocal novel adjacencies that involves the adjacent extremities jh and (j + 1)t must have been created during a single rearrangement event that broke both the reference adjacency {jh, (j + 1)t} as well as reference adjacencies involving x and y. Thus, we identify evidence of complex rearrangements by finding chains of novel adjacencies where consecutive novel adjacencies, ai and ai+1, contain reference-adjacent extremities (Fig. 5A). Such a chain provides a lower bound on the number k of simultaneous DNA breakages that must have taken place, that is, it is possible that a rearrangement with k ≥ 3 double-stranded DNA breaks may not have produced novel adjacencies connecting all of the k broken reference adjacencies.
Evidence of complex k-break (k ≥ 3) rearrangements in metastatic prostate cancer. (A) Two complex rearrangements across two genomes in a heterogeneous sample. A 5-break rearrangement that produced four novel adjacencies {a, b, c, d} involving five reference adjacencies (X, R, L, O, and M), with novel adjacency a not present in genome G2. A 3-break rearrangement that produced three novel adjacencies {e, f, j} involving three reference adjacencies (Y, Z, and T), with novel adjacency j not present G1. (B, top) A complex 5-break rearrangement on Chromosome 10 in the karyotype inferred by RCK on sample A31a. Only the four novel adjacencies, five reference adjacencies, and incident segments involved in the rearrangement are shown. Copy numbers ≤1 are omitted for clarity, and absent segments/adjacencies are shown as faded. (Bottom) The locations of the corresponding double-stranded DNA breakages for the 5-break on Chromosome 10, indicated as x|y for each reference adjacency {(x)h, (y)t}. Three reference adjacencies lie in/near genes: reference adjacency 102,756,[799|800] falls within the promoter region for gene LZTS2; reference adjacency 114,208,50[2|3] falls inside gene VTI1A; and reference adjacency 114,062,94[6|7] falls inside gene TECTB. (C) Number of complex k-break (k ≥ 3) rearrangements reported in RCK-reconstructed karyotypes using HATCHet and Battenberg copy number inputs with novel adjacency utilization parameter P = 0.9. Values of 0 are omitted for clarity.
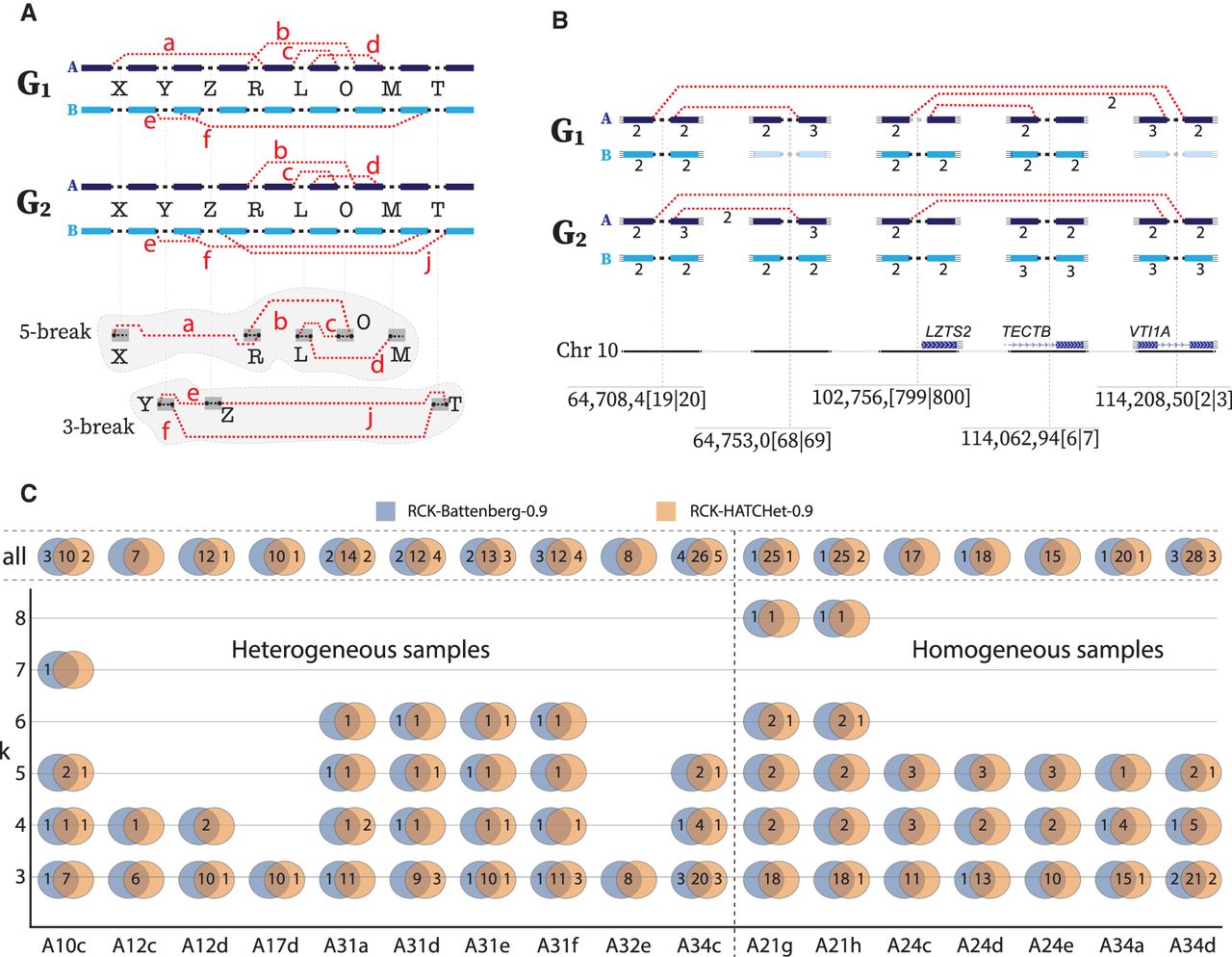
We identified complex k-break rearrangements in the karyotypes reconstructed by RCK (P = 0.9) on all 17 metastatic prostate cancer samples. One example is a 5-break on Chromosome 10 in heterogeneous sample A31a (Fig. 5B). All five novel adjacencies resulting from the 5-break are present in one of the genomes inferred by RCK, while only 4/5 of the novel adjacencies are present in the other (subclonal) genome. Moreover, the copy numbers for some of these novel adjacencies differ across the two genomes, suggesting that additional subclonal rearrangements occurred after the complex rearrangement. Three of the reference adjacencies affected by this 5-break fall within the genes VTI1A, TECTB, and LZTS2 that are listed in the COSMIC database of genes somatically mutated in cancer (Forbes et al. 2017). Note that some extremities involved in reciprocal novel adjacencies have no change in copy number (e.g., Chr10:64,708,4[19|20] in genome G1), whereas others show a change in copy number (e.g., Chr10:114,208,50[2|3] in genome G1) (Fig. 5B). These observations underscore the importance of RCK’s karyotype reconstruction model that allows for genomic heterogeneity within a sample and also carefully analyzes reciprocal novel adjacencies.
Overall, the number of k-breaks identified by RCK ranges from seven (in A12c, all 3-breaks) to 31 (in A34d, a mixture of 3-, 4-, and 5-breaks) per sample. Moreover, these k-breaks showed strong concordance between HATCHet and Battenberg segment copy number input (Fig. 5C). The most frequent complex rearrangements were 3-break rearrangements, which were present in all samples. We also find two 8-break rearrangements, one each in samples A21g and A21h from patient A21. We found that 266/302 (respectively, 260/296) of complex rearrangements overlapped human genes from RefSeq (O'Leary et al. 2016) using the karyotypes inferred by RCK with HATCHet (respectively, Battenberg) segment copy number inputs (Supplemental Table S5). Of these genes, 14 (respectively, 15) are in the COSMIC. In total, we identified 185 distinct genes being affected by complex rearrangements ranging from eight (in sample A12c) to 33 (in sample A34c) per sample.
Discussion
We introduced RCK, a novel algorithm for reconstructing clone- and haplotype-specific cancer genome karyotypes from bulk tumor samples. RCK accounts for heterogeneity in the observed tumor sample, correctly models the diploid reference genome, and enforces biologically reasonable evolutionary constraints that generalize the infinite sites assumption to somatic large-scale genome rearrangements. RCK is, to the best of our knowledge, the only algorithm with these features and also the only algorithm that can combine both second- and third-generation sequencing data into the reconstruction process, leveraging the long-range adjacency information from third-generation sequencing technologies.
On prostate cancer sequencing data, we found that RCK infers cancer karyotypes whose inferred segment copy numbers are closer to those produced by state-of-the-art copy number inference tools (HATCHet and Battenberg), and whose novel adjacencies conform with constraints from an infinite sites evolutionary model. In contrast, ReMixT’s approach of using novel adjacencies to “adjust” copy numbers generally led to allele-specific segment copy numbers that were different from those of HATCHet and Battenberg. Moreover, the novel adjacencies that are present in ReMixT inferred karyotypes often require biologically implausible rearrangements. Furthermore, we identified complex k-break rearrangements in all but two of the prostate cancer samples, which overlap a total of 185 genes, including known cancer genes in COSMIC. These results show that RCK’s proper handling of reciprocal novel adjacencies plays a crucial role in adequate reconstruction of clone- and haplotype-specific cancer karyotypes.
Although RCK uses a comprehensive somatic evolutionary model and addresses some shortcomings of the previous approaches, there are several limitations and avenues for future improvement. First, RCK’s performance is limited by the allele-specific copy numbers and novel adjacencies provided in input. Our analysis of prostate tumors focused on the samples for which two copy number deconvolution methods (HATCHet and Battenberg) agreed about the heterogeneous composition of the sample. Further improvements in methods for copy number deconvolution will increase RCK’s accuracy in deriving allele- and haplotype-specific karyotypes. RCK can use output from different methods for inferring allele-specific copy numbers from bulk samples; however, nearly all such current methods require a matched normal sample, and thus in practice RCK also requires a matched normal sample. Second, improvements in distinguishing novel telomeres (telomeres not present in the reference genome) from errors in the input data remains a challenging problem. In the RCK results presented here, we assume that each cancer genome contains only the telomeres present in the reference genome, that is, no new telomeres are present in the cancer genomes. Although the current implementation of RCK allows the user to specify the location of novel telomeres, telomere selection is not currently part of the objective function optimized by RCK. Third, additional work is needed to distinguish karyotypes with haplotype symmetries that produce identical values of RCK’s objective function. RCK uses reciprocal locations and input allele-specific profiles to assign haplotypes, but these constraints are usually insufficient to infer chromosome-scale haplotypes. Additional information indicating that multiple novel adjacencies should be assigned to the same haplotype in the karyotype would reduce ambiguities in karyotype inference and provide longer resolved haplotype blocks; for example, Aganezov et al. (2020) used RCK’s model with additional haplotype label constraints arising from the third-generation long reads. Fourth, some extremely rearranged cancer genomes may violate the generalized infinite sites constraints and have extremities that are involved in several distinct unlabeled novel adjacencies. Such instances can be identified from the input novel adjacencies and the current implementation of RCK allows users to explicitly specify location(s) where such breakpoint reuse is suspected, thus providing a manual control over the extremity-exclusivity constraint. It would be interesting to extend this approach to evaluate and score cases in which the data support breakpoint reuse. Fifth, we can further generalize RCK to simultaneously analyze multiple samples from the same individual, as has proven useful in copy number inference (Zaccaria and Raphael 2020). Extensions to multiple samples could leverage phylogenetic (Zaccaria et al. 2017), spatial (El-Kebir et al. 2018), or temporal (Myers et al. 2019) relationships between samples. Finally, it would be helpful to incorporate a patient-specific germline genome to better distinguish germline structural variations, long repetitive segments, and so forth.
Higher-resolution reconstructions of cancer karyotypes can help researchers illuminate differences/similarities between different types of cancer in general and lead to a more targeted and personalized medical treatments in specific patients. RCK’s inference of clone- and haplotype-specific cancer karyotypes enables further studies of the somatic mutational processes that produce highly rearranged cancer genomes, as well as improved characterization of specific functional changes (e.g., loss of heterozygosity, novel haplotype-specific fusion genes, and so forth).
Methods
We derive the RCK algorithm by first formulating the mathematical problem in the case of “perfect” input data (“Sequencing of rearranged cancer genomes”). Next, we describe the construction of the diploid interval adjacency graph (“Diploid interval adjacency graph”) and show how the model can incorporate information from third-generation sequencing technologies (“Third-generation sequencing technologies and novel adjacency groups”). Finally, we describe the realistic case when there is uncertainty in input segment copy number values (“Uncertainty in copy number measurements”), and formulate the optimization problem solved by RCK.
Sequencing of rearranged cancer genomes
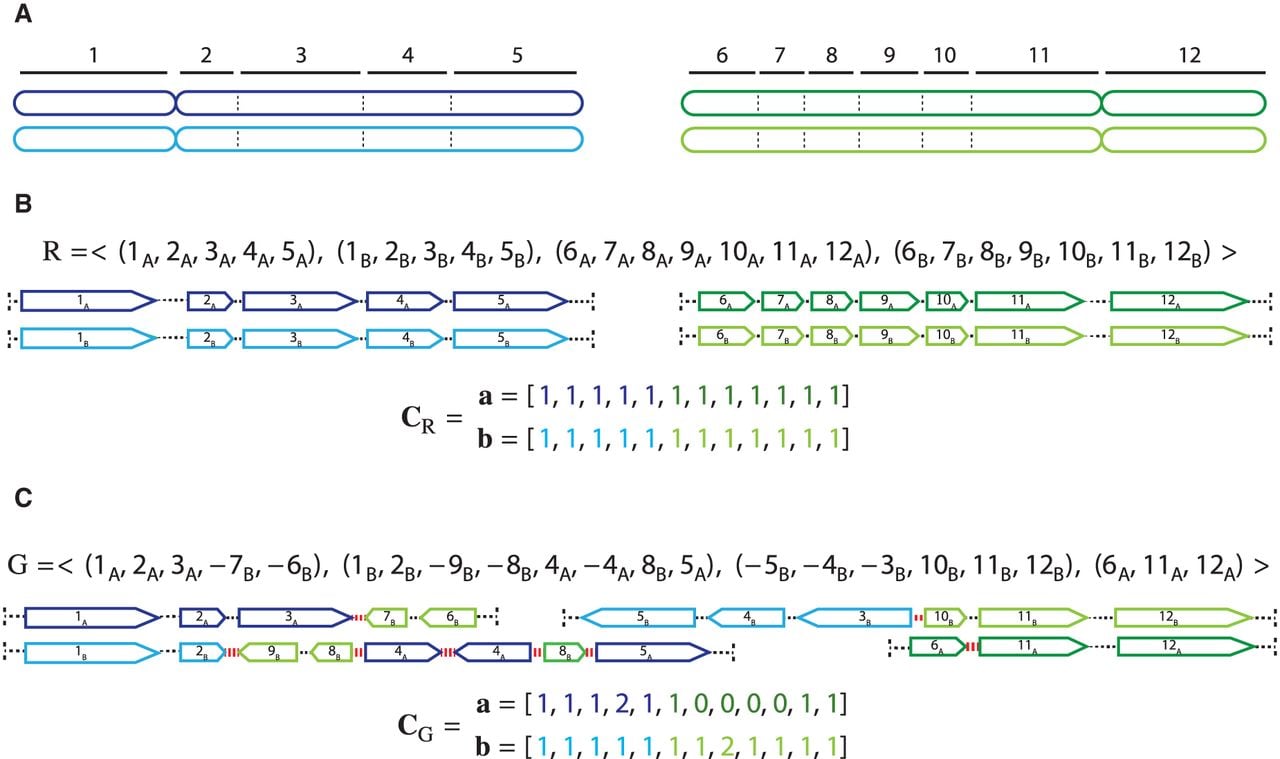
A cancer genome results from a sequence of somatic genome rearrangements that transform the diploid reference human genome R into a derived genome G. In general, tumors are genetically heterogeneous with cancerous cells distinguished by unique rearrangements. Sequencing of a bulk tumor sample thus measures not a single derived genome, but rather a mixture of different derived genomes, often called clones. We define a sample to be a list of n derived genomes all of which were derived from the same diploid reference genome R via large-scale rearrangements.
The genome rearrangements that produce each derived genome result in duplications, deletions, and reorderings of segments of the reference genome. Thus, we describe each derived genome using an “alphabet” of segments of the reference genome. Every chromosome in a diploid reference genome R is present in two homologous copies, which we label by A and B, respectively. Given a multichromosomal diploid reference genome R, we label segments 1 through m (Fig. 6A). A segment is a contiguous part of the H homolog, H ∈ {A, B} of a reference chromosome; the endpoints and are called extremities. In a derived genome, segments can be absent, present more than once, and appear both in forward and reverse orientation. We denote by a reversed instance of the segment . Extremities that demarcate the beginning and the end of a chromosome are called telomeres, and we define by the set of telomeres in genome G. For a diploid reference genome R with k chromosomes, we define the set of reference telomeres and note that .
A derived genome G corresponds to a collection of derived chromosomes, where each derived chromosome is a concatenation of segments from any homologous copy of any of the chromosomes in the diploid reference R. Each derived chromosome thus corresponds to a word from the following alphabet:
A genome G determines a diploid segment copy number profile , where values (aj, bj) ∈ ℕ2 indicate the number of copies of segments and in G, respectively (Fig. 6C). Note that a diploid reference genome R has aj = bj = 1 for every segment j. Similarly, a sample determines a pair of n × m diploid segment copy number matrices, where genome-specific segment copy number vectors and contain integer values ai,j, bi,j ∈ ℕ corresponding to the number of times segments and appear in genome Gi ∈ S, respectively. We denote by and by vectors of copy number values for segments and across all genomes Gi ∈ S.
Segments, extremities, and copy number profiles for genomes. (A) A diploid reference genome r containing two pairs of homologous chromosomes: A Chromosomes are dark blue and dark green, and the homologous B Chromosomes are light blue and light green. Chromosomes are partitioned into consecutive segments labeled 1 through 12. (B, top) Reference genome r is a collection of concatenations of segments; the “flat” end of segment j corresponds to the tail extremity jt, whereas the “pointy” end of each segment j corresponds to the head extremity jh. Dashed lines correspond to reference adjacencies between adjacent extremities. The set of extremities is the telomere set. (Bottom) The diploid segment copy number profile CR = (a, b) for the genome R with colors (dark/light blue/green) corresponding to A/B labeled segments. (C, top) A derived genome g obtained via multiple large-scale rearrangements from the reference genome R. Red dashed lines correspond to novel adjacencies, for example, . (Bottom) The diploid segment copy number profile CG = (a, b) for the genome g with colors (dark/light blue/green) corresponding to A/B labeled segments. The set of telomeres in the derived genome G is identical to the set of telomeres in the reference genome R.
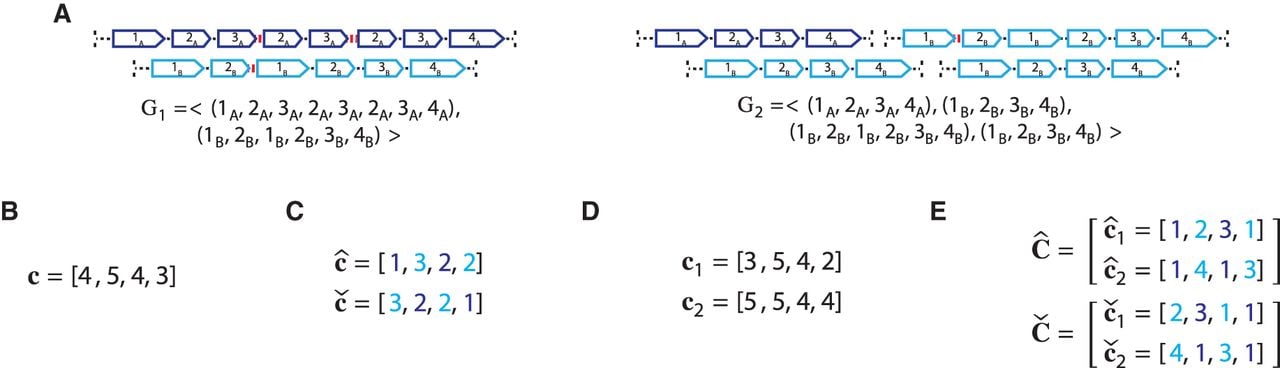
Current short-read sequencing technologies do not measure the diploid segment copy number profile CG of a derived genome G directly. Rather there exist multiple methods (Van Loo et al. 2010; Boeva et al. 2012; Carter et al. 2012; Nik-Zainal et al. 2012; Fischer et al. 2014; Ha et al. 2014; McPherson et al. 2017; Zaccaria and Raphael 2020) that derive a pair , of vectors, where for every segment j an unlabeled (allele-specific) pair represents copy numbers of segments and in G, but without A/B labels explicitly associated with the measured values. In other words, we know that , but it is unclear whether or (example shown in Fig. 7). Similarly for a sample , we do not measure the pair CS = (A, B) of its n × m diploid segment copy matrices directly, but rather we measure a pair of n × m allele-specific segment copy number matrices, such that for every segment j either or . Figure 7 shows examples of copy number profiles for a heterogeneous sample (Fig. 7A) derived under different assumptions about the sample (e.g., haploid [Fig. 7B] vs. diploid reference [Fig. 7C], homogeneous vs. heterogeneous sample [Fig. 7D,E]).
Ambiguity and errors in inferring segment copy number (SCN) profiles for a heterogeneous sample S = (G1, G2) under different assumptions about the sample composition. (A) A two-genome proper sample S = (G1, G2): each genome Gi ∈ S is depicted as collections of adjacent blocks (top), and the corresponding sequences of signed blocks (bottom). (B) The copy number profile c = [c1, c2, c3, c4] inferred under the assumption that the sample is homogeneous (i.e., comprised of a single derived genome) and the reference genome is haploid (i.e., each segment has only a single haplotype in the reference). Each value cj is the weighted average of the sums of haplotype-specific (or allele-specific) copy numbers over the genomes Gi ∈ S. (C) Allele-specific copy number profiles and inferred under the assumption that the sample is homogeneous and the reference genome is diploid (i.e., each segment has two haplotypes labeled A and B). Here, the entries and for segment j are averages and of genome- and allele-specific copy number values. Note that the vectors and č do not preserve the true A/B label of each allele: dark blue are true counts of allele A and light blue are true counts of allele B. Here, segments 2 and 4 are flipped. (D) Genome-specific copy number profiles c1 = [c1,1, c1,2, c1,3, c1,4] and c2 = [c2,1, c2,2, c2,3, c2,4] inferred under the assumption that the sample is heterogeneous, but the reference genome is haploid. Here, the entry ci,j for a segment j and genome Gi is the sum of allele-specific copy number values in a genome Gi. (E) Allele- and genome-specific copy number matrices inferred under the assumption that the sample is heterogeneous and the reference genome is diploid. Segments 2 and 4 are flipped alleles: and .
In addition, current short-read sequencing technologies do not measure the set of adjacencies in a genome G derived from a diploid reference R; rather for every novel adjacency we measure only an unlabeled adjacency where the extremities are missing the A/B labels. For example, in the derived genome G shown in Figure 6, we measure the unlabeled novel adjacency {3h, 7h} instead of the true novel adjacency . There exist several methods capable of producing the unlabeled novel adjacencies both from a standard short-read bulk sequencing data (Rausch et al. 2012; Sindi et al. 2012; Layer et al. 2014; Sudmant et al. 2015; Chen et al. 2016; Wala et al. 2018) as well as from third-generation sequencing technologies (English et al. 2014; Ritz et al. 2014; Zheng et al. 2016; Huddleston et al. 2017; Spies et al. 2017; Elyanow et al. 2018; Nattestad et al. 2018; Sedlazeck et al. 2018). Similarly for every novel adjacency in sample , we measure only the unlabeled counterpart . Moreover, we also lose the information about which genome(s) in sample S contains the novel adjacency a. We define by a set of unlabeled adjacencies measured from a sample S.
Because the measured unlabeled novel adjacencies do not include the A/B labels, we do not know the true underlying novel adjacencies that produced a measurement. For an unlabeled novel adjacency , let be the set of the four possible novel adjacencies that can be obtained by labeling extremities in a with haplotypes A or B. For a set A of unlabeled novel adjacencies, let be the set of all possible labeled novel adjacencies that can be obtained from A. Note that when a set of unlabeled novel adjacencies comes from a genome G, it follows that . Then the set gives all possible adjacencies that can be present in the genome G.
Reconstructing karyotypes under generalized infinite sites constraints
To reconstruct cancer genomes using the ambiguous measurements of adjacencies and copy number profiles that are obtained from bulk sequencing data, we make some simplifying assumptions on the somatic evolution process that generated the genomes Gi in samples S. Specifically, we make the infinite sites assumption that the large-scale somatic rearrangements that “break” and “glue” chromosomes do not affect the exact same nucleotide more than once during evolution. Previous work on genome rearrangements has used the infinite sites assumption (Ma et al. 2008; Alekseyev and Pevzner 2009; Oesper et al. 2012; Li et al. 2016; McPherson et al. 2017); however, in the case of a diploid reference genome, there are multiple possible interpretations of the infinite sites assumption that depend on whether different rearrangements at homologous nucleotide positions are allowed. We define several specific assumptions about reuse of extremities and adjacencies that collectively we refer to as the generalized infinite sites assumptions.
Specifically, we assume that the large-scale somatic rearrangements that “break” and “glue” chromosomes do not affect the same genomic location—on either of the two homologous chromosomes A and B in any of the genomes Gi in the sample S—more than once during the entire somatic evolutionary process. This generalized infinite sites assumption leads to the following constraints on the extremities and adjacencies of each derived genome G:
a. Extremity-exclusivity: Every extremity is involved in at most one novel adjacency from . This constraint derives from the fact that a novel adjacency that includes the extremity results from a large-scale rearrangement that breaks a reference adjacency that includes (and possibly several other reference adjacencies). By the generalized IS assumptions, at most one novel adjacency can involve the extremity .
b. Homologous-extremity-exclusivity: If an extremity is involved in a novel adjacency from , then the homologous extremity is not involved in any novel adjacency from . This constraint follows the logic of the extremity-exclusivity constraint, but further restricts rearrangements to involve at most one of the homologous extremities and . This constraint derives from the fact that for both extremities and to be involved in novel adjacencies, there must have been at least two large-scale rearrangements breaking homologous reference adjacencies involving both extremities and , which is prohibited under the generalized IS.
c. Homologous-reciprocal-extremity-exclusivity: If an extremity from the reference adjacency is involved in a novel adjacency from , then the homologous extremity is not involved in any novel adjacency from . This constraint follows the observation that for both extremities and to be involved in novel adjacencies, there must have been two large-scale rearrangements breaking both homologous reference adjacencies and , which is prohibited under the generalized IS.
Supplemental Figure S11 gives examples of rearrangements that violate the generalized IS and the resulting implications for novel adjacencies in the derived genomes.
We call a genome G proper provided that the extremity-exclusivity, homologous-extremity-exclusivity, and homologous-reciprocal-extremity-exclusivity constraints hold for the set . Similarly, we call a sample proper if the three constraints hold for the set of novel adjacencies in all of the genomes in S. Thus, the generalized IS constraints are imposed for the whole somatic evolutionary process that produced the genomes in the sample S. Note that if a sample is proper, then any subsample (including individual derived genomes Gi ∈ S of S is also proper. Note that if a set of unlabeled novel adjacencies is measured from a proper sample S, then satisfies the generalized IS conditions. This is because unlabeled novel adjacencies involve extremities that lack A/B labels, and thus only the (unlabeled) extremity-exclusivity constraint (i.e., on unlabeled extremities) must be satisfied. This is achieved because in a proper sample, the extremity-exclusivity and homologous-extremity-exclusivity conditions guarantee that for every pair of homologous extremities at most one of them is involved in any novel adjacency from , and thus the unlabeled extremity is also involved in at most one measured unlabeled novel adjacency from .
We assume that large-scale rearrangements that generated a derived genome G from a diploid reference genome R have not created novel telomeres (i.e., ), and formulate the following problem of reconstructing a sample of derived genomes from measurement data.
Cancer Genome(s) Reconstruction Problem
Given a diploid reference genome R, a pair of n × m allele-specific segment copy number matrices, and a set of measured unlabeled novel adjacencies that satisfies (unlabeled) extremity-exclusivity constraint, find a proper sample such that:
for every adjacency , either or ;
for every adjacency , there exists a unique pair H, H′ ∈ {A, B} of labels, such that ;
for every segment j, either or ;
for every genome Gi ∈ s, the telomere set .
Diploid interval adjacency graph
We reformulate the Cancer Genome(s) Reconstruction Problem as a graph-theoretic problem, which provides a convenient framework for deriving a combinatorial optimization algorithm to solve the problem. First, we define the diploid interval adjacency graph (DIAG), a graph that summarizes the segments, extremities, and adjacencies of a genome, or genomes, derived from the diploid reference genome. The DIAG can be viewed as a generalization of a breakpoint graph used in the area of comparative genomics (Alekseyev and Pevzner 2009; Avdeyev et al. 2016; Zerbino et al. 2016), or graphs used in the area of structural analysis of normal and cancer genomes with haploid reference structure (Medvedev et al. 2010; Oesper et al. 2012; Li et al. 2016; Dzamba et al. 2017; Eitan and Shamir 2017; McPherson et al. 2017).
A DIAG is constructed on a set of segments, and a set of adjacencies (Fig. 8). The set V of vertices is in one-to-one correspondence with all segments’ extremities. Formally we define V as follows:
A DIAG constructed on a set of segments, and a set of adjacencies. The set corresponds to reference adjacencies in a diploid reference R shown in Figure 6B, and the set represents unlabeled novel adjacencies that were measured from a derived genome G shown in Figure 6C. Squares indicate telomere vertices , and circles are non-telomere vertices. Solid edges correspond to segment edges in ES, with dark blue/green edges corresponding to segments labeled A, and light blue/green edges corresponding to segments labeled B. Black-dashed edges are reference adjacency edges ER, and red-dotted edges are novel adjacency edges EN.
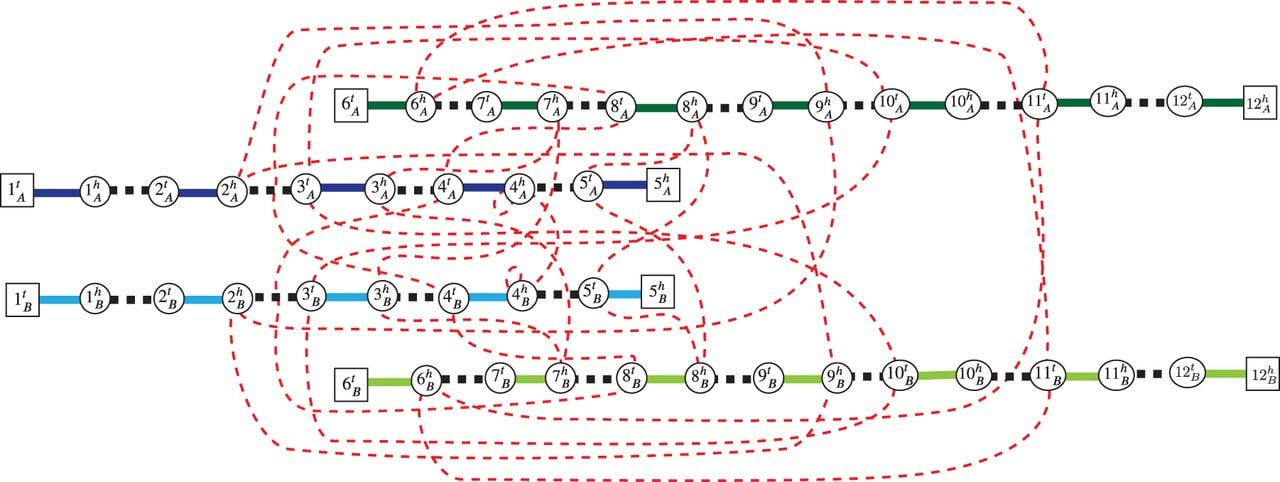
Because every vertex is incident to exactly one segment edge , we define eS(v) ∈ ES to be a segment edge incident to a vertex v, and define eS(jH) ∈ ES to be a segment edge corresponding to a segment jH. Every vertex v ∈ V is incident to at most one reference adjacency edge, and we define eR(v) ∈ ER to be the reference adjacency edge containing vertex v, if such adjacency exists. We define EN(v)⊆ EN to be the set of novel adjacency edges incident to v ∈ V.
Every chromosome in a derived genome G determines a segment-adjacency edge alternating walk in the corresponding DIAG that starts and ends at telomere vertices in (Supplemental Fig. S12B). Such an alternating walk spells out a concatenation of segments from the reference genome, corresponding to a derived chromosome in G. Thus, a derived genome G determines a collection of segment-adjacency edge alternating walks. The number of times a segment edge is traversed (in either direction) across all walks determined by G corresponds to the segment copy number (e.g., ). Similarly, the number of times an adjacency edge is traversed (in either direction) across all walks determined by G corresponds to an adjacency copy number (i.e., the number of times an adjacency corresponding to an edge e is present in G). A genome G thus determines an edge multiplicity function μ:E → ℕ on both segment and adjacency edges (example is shown in Supplemental Fig. S12A). We call the corresponding DIAG a weighted DIAG.
We define by an auxiliary function that outputs 2 if a is a self-loop adjacency (edge), and 1 otherwise. For a genome Gi in a sample S, a vertex v ∈ V exhibits copy number balance provided
The following theorem follows directly from previous work (Kotzig 1968; Pevzner 1995):
A weighted DIAG D = (V, E, μ), can be partitioned into a collection of segment-adjacency edge alternating walks that start and end at a set of telomere vertices, such that every edge e ∈ E is traversed μ(e) times provided:
Every non-telomere vertex is copy number balanced;
Every telomere vertex has copy number excess.
When the derived genome is allowed to have circular chromosomes, which have been extensively observed and studied in cancer (Carroll et al. 1988; Von Hoff et al. 1988; Fan et al. 2011; Garsed et al. 2014; Turner et al. 2017), Theorem 1 provides not only a necessary but also a sufficient condition for a derived genome to exist. An extended discussion about DIAG decomposition into segment-adjacency edge alternating walks is in “On minimal path-cycle Eulerian decomposition of (D)IAGs” in Supplemental Methods.
For every unlabeled novel adjacency and a DIAG , we define by hE(a)⊆ EN a subset of novel adjacency edges corresponding to adjacencies in h(a). Furthermore, given a weighted DIAG , for every unlabeled novel adjacency we define by a subset of adjacency edges with positive multiplicities as follows:
For every unlabeled adjacency and a genome Gi ∈ S, we define by a subset of novel adjacency edges in hE(a) with positive copy number as determined by the genome-specific edge multiplicity function μi as follows:
We reformulate the Cancer Genome(s) Reconstruction Problem into a problem of finding edge multiplicity functions μ1, μ2, …, μn:E → ℕ in the corresponding DIAG as follows.
Cancer Karyotype Reconstruction Problem (Exact Data)
Given a DIAG , where the set of unlabeled novel adjacencies satisfies the (unlabeled) extremity-exclusivity constraint, and a pair of n × m allele-specific segment copy number matrices, find edge multiplicity functions μ1, μ2, …, μn:E → ℕ such that:
for every adjacency , ;
for every i ∈ [n] and every adjacency , ;
for every pair of unlabeled novel adjacencies, such that , there exists and , where H, H′, H′′ ∈ {A, B};
for every segment j, either or ;
for every i ∈ [n] and every non-telomere vertex the copy number balance condition (equality (5)) holds;
for every i ∈ [n] and every telomere vertex either the copy number balance condition (Eq. (5)) or the copy number excess condition (Eq. (6)) holds.
Edge multiplicity functions μi that solve the above problem guarantees the existence of a proper sample S defined by the μi; however, the solution may not be unique, with several solutions achieving the same objective function value and satisfying the required constraints. Specifically, if μ1, μ2, …, μn are a solution, then every segment j satisfying the following two conditions defines a symmetrical counterpart solution: (1) normal adjacencies defined by the μi do not involve/span extremities of either jA or jB; and (2) the edge multiplicities of segment j are haplotype-symmetric: . A symmetrical counterpart solution can be obtained from a solution by flipping the A/B haplotype labels of all segment and adjacency copy numbers on one side of the chromosome that contains a segment j satisfying the two conditions given above (Supplemental Fig. S13).
Third-generation sequencing technologies and novel adjacency groups
Recently, several third-generation sequencing technologies have been introduced including single-cell DNA sequencing, barcoded linked reads, and long-read sequencing (English et al. 2014; Ritz et al. 2014; Zheng et al. 2016; Huddleston et al. 2017; Spies et al. 2017; Elyanow et al. 2018; Sedlazeck et al. 2018). These technologies can provide additional information about groups of novel adjacencies that are present in the same genome. For example, a single-cell sequencing can reveal that several novel adjacencies are present on the same derived genome, and long-read sequencing can reveal that multiple novel adjacencies are present on the same DNA molecule. We define a molecule group to be a set of unlabeled novel adjacencies that originate from a single derived genome Gi ∈ S. Recall that for every unlabeled novel adjacency measured in a sample S = (G1, G2, …, Gn) there exists a unique novel adjacency , when S is proper. Or, more formally,. Similarly, for every molecule group of unlabeled novel adjacencies obtained from third-generation sequencing of a proper sample S = (G1, G2, …, Gn), there is at least one genome Gi ∈ S such that
Let U denote the set of molecule groups obtained from a third-generation sequencing experiment. Below we extend the Cancer Karoytype Reconstruction Problem to leverage information provided by U.
Uncertainty in copy number measurements
Inferring allele-specific segment copy number matrices from bulk sequencing is challenging, and existing inference methods do not infer these copy numbers without error. In addition, novel adjacencies further subdivide the genome segments output by allele-specific copy number methods, as described in “Deriving extremities and novel adjacencies from data” below. We formulate the Cancer Karotype Reconstruction Problem to address these ambiguities.
First, we derive a distance between copy number matrices that accounts for both allele-flipping—owing to uncertainty in haplotype labels—and fragmentation of copy number segments. Formally, we define a fragment f[j,l] to be a sequence (j, j + 1, …, j + l) of segments that are adjacent on the reference genome. We denote by F a collection of nonoverlapping fragments that cover all of the segments. The allele-specific copy numbers given in input define the number of copies of each allele for all segments within a fragment. We aim to leverage this information about correlations between allele-specific copy numbers for segments from the same fragment when we infer segment copy number values. Given a pair of n × m allele-specific segment copy number matrices, a pair C = (A, B) of n × m haplotype-specific segment copy number matrices, and a set F of fragments, we define a copy number distance
We now extend the Cancer Karyotype Reconstruction Problem to the case edge multiplicity functions on the edges of the corresponding DIAG where the measured allele-specific segment copy numbers are noisy and where (optionally) a set U of molecule groups from third-generation sequencing is available.
Cancer Karyotype Reconstruction Problem
Given a DIAG , where the set of unlabeled measured novel adjacencies satisfies (unlabeled) extremity-exclusivity constraint, a pair of n × m allele-specific segment copy number matrices, a set F of fragments, and (optionally) a set U of molecule groups of unlabeled novel adjacencies, find edge multiplicities functions μ1, μ2, …, μn:E → ℕ such that:
conditions 1–6 of the Exact Data problem are satisfied;
for every molecule group there exists (at least one) i (1 ≤ i ≤ n) such that ;
the copy number distance is minimized for a pair of diploid segment copy number matrices determined by values of edge multiplicity functions μ1, μ2, …, μn on segments edges ES.
In the Supplemental Methods, we derive a mixed-integer linear program (MILP) optimization problem that solves the Cancer Karyotype Reconstruction Problem. We find that the run times of RCK are reasonable, requiring on average <15 min on simulated data (Supplemental Fig. S14) and <10 min on most of the prostate cancer samples (Supplemental Fig. S15).
Simulating rearranged cancer samples
We simulated 100 instances of a cancer samples S = (G1, G2), containing two clones with corresponding genomes G1 and G2. In each instance, G1 and G2 share the majority of the somatic rearrangement history (∼600 rearrangements), but also have clone-specific rearrangements affecting their structure (100 and 200 simple rearrangements for clones G1 and G2, respectively). For every generated cancer sample S = (G1, G2) we also created a homogeneous cancer sample S′ = (G1) containing the first cancer clone in S. For each sample S, we derived the true clone-specific segment copy number profile CS and the true set of novel adjacencies. To simulate ambiguity in deriving segment copy numbers and novel adjacencies from DNA sequencing data, we remove the clone label for each novel adjacency and the haplotype labels for each segment copy number and novel adjacency. We refer to this process as clone and haplotype information loss (CHIL) (Supplemental Fig. S1C). First, we generate CHIL segment copy number input by randomly (with probability of 0.5) allele-flipping copy numbers in C for every segment j. We then generate CHIL novel adjacencies by removing haplotype labels on extremities in novel adjacencies in .
In real data, errors in novel adjacencies and clone- and allele-specific segment copy numbers are expected. To simulate such errors, we first simulated a fixed-size fragment-averaged copy number input by averaging true segment copy numbers from C over 50 kbp fragments. We then simulated noisy novel adjacency measurements by varying the coordinates of segment extremities by ±50 bp in a random half of adjacencies from , and then lastly generating an additional 10% of spurious novel adjacencies, resulting in a noisy set of input novel adjacencies. We then performed the same CHIL procedure on and to obtain the inputs and for RCK (Supplemental Fig. S1C). We also simulate errors in input copy number data by perturbing the copy numbers by ±1 in a random 5% of the segments before performing the CHIL procedure, resulting in segment copy number input for RCK.
Deriving extremities and novel adjacencies from data
We derive the extremities and novel adjacencies that form the input to RCK by integrating the output from structural variant prediction methods and copy number inference methods as follows. Structural variant prediction methods output novel adjacencies (sometimes called breakpoints) in the form of pairs: {(chr1, coord1, str1), (chr2, coord2, str2)}, where chri determines the chromosome of origin the genomic loci i, coordi determined the coordinate of the genomic loci i on the respective chromosome chri, and determined the strand of origin of the genomic loci i. Methods to predict allele-specific copy numbers partition the reference genome into nonoverlapping fragments F. The challenge in combining these two outputs is that the coordinates (extremities) output by a structural variant prediction method and an allele-specific copy number method are often not identical. This is because most methods to predict structural variants do not predict breakpoints to single-nucleotide resolution, and thus there is some uncertainty in the exact values of the coordinate coordi of the genomic loci i involved in a novel adjacencies. This uncertainty can be an issue when determining whether an adjacency is part of a reciprocal event (e.g., inversion or reciprocal translocation). Similarly, methods to compute allele-specific copy numbers often have uncertainty in the genomic locations where changes in copy number occur.
First, we refine the positions of extremities involved in reciprocal novel adjacencies. For a sample S, we sort the positions involved in unlabeled novel adjacencies from on every chromosome. Then, we use a sliding window to update the coordinates for any consecutive pair pi, pj of positions which resembles a reciprocal signature, that is, if the distance was <50 base pairs and , we update the values of the coordinates in positions pi and pj so that they have a coordinate distance of 1, with the position having a + strand appearing before the position having a − strand (Fig. 9A).
Derivation of extremities and novel adjacencies for input to RCK and ReMixT. (A) An example of derivation of coordinates that resembles a reciprocal signature in measured unlabeled novel adjacencies on a chromosome a. Positions p1 = (a, 100, + ) and p2 = (a, 107, − ) have reciprocal signature (i.e., and ). Updated pair of coordinates constitutes a reciprocal location. (B) An example of partitioning of a set of fragments from allele-specific copy number calls into a set S = {s1, s2, s3, s4, s5, s6, s7, s8} of segments. Extremities of segments in S correspond to either preprocessed coordinates of unlabeled novel adjacencies (e.g., ,) or to the extremities of fragments in F (e.g., ,).
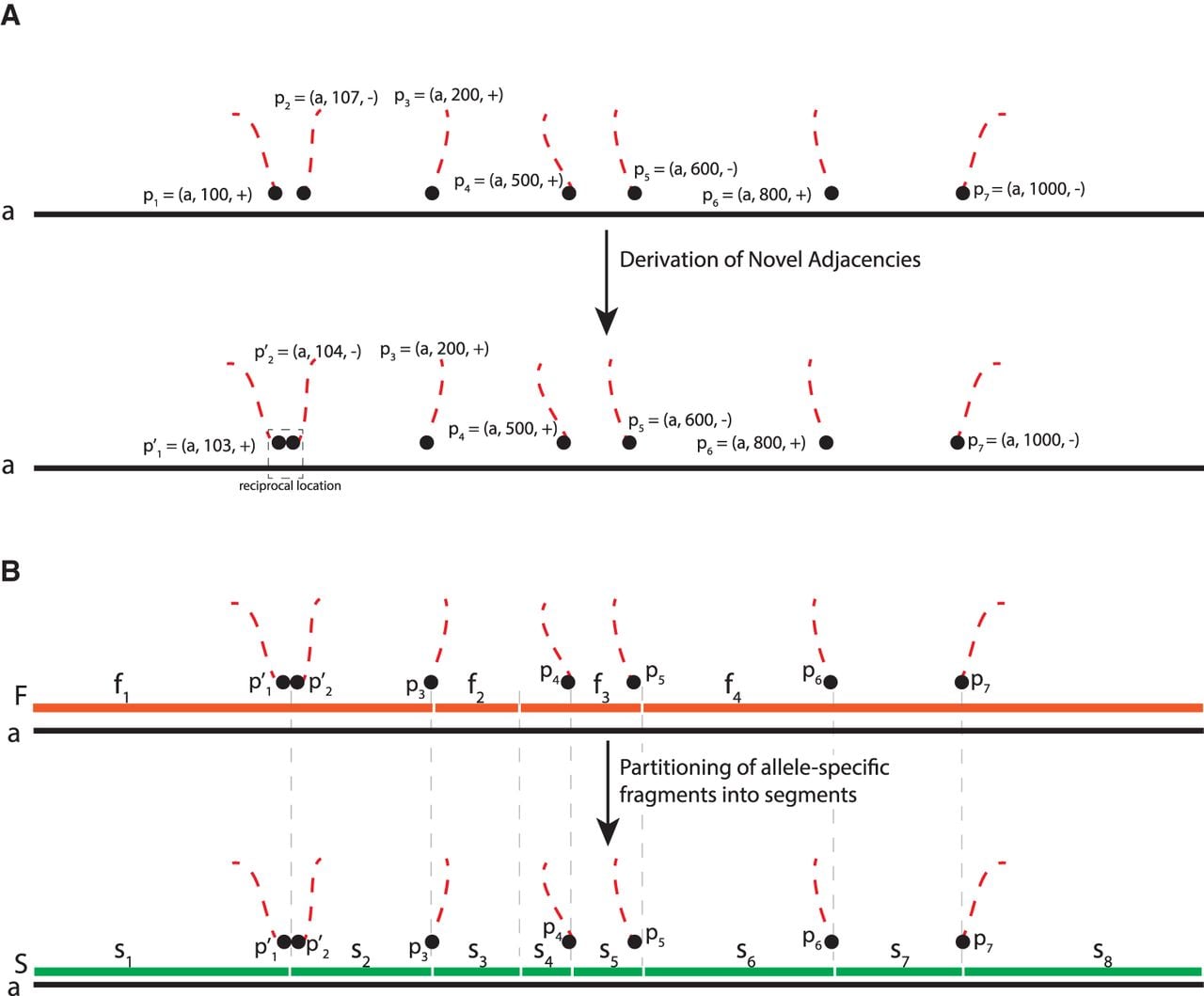
Next, we adjust the extremities identified by structural variation predictions and allele-specific copy number segmentation as follows. We partition the fragments F, on which allele-specific copy number values are measured, into smaller segments [1, 2, …, m] such that extremities of these segments correspond either to the coordinates of extremities involved in the refined novel adjacencies from or to the extremities of the original fragments (Fig. 9B). This results in original fragments from F spanning one or more smaller refined segments. Copy numbers on the newly obtained segments are inherited from the values of the “parent” spanning fragments.
In the analysis of the prostate cancer data set (Gundem et al. 2015), we used novel adjacencies from the original publication, which were obtained using brass2 (https://github.com/cancerit/BRASS). For allele-specific copy numbers, we used the output from Battenberg from the original publication (Gundem et al. 2015). We also inferred clone- and allele-specific copy numbers using HATCHet (Zaccaria and Raphael 2020), which we ran on the read alignments obtained from the original publication. For each sample, the output of Battenberg and HATCHet includes (1) the number of clones; (2) allele-specific copy numbers for each genomic segment in each clone; and (3) the occurrence of a whole-genome duplication (WGD) when reported tumor ploidy is >3. Summary statistics for these samples is available in Supplemental Table S6.
Last, to compute length-weighted segment copy number distances between RCK, ReMixT, Battenberg, and HATCHet on the prostate cancer samples, we refined the fragments/segments on which the copy numbers were inferred as shown in Supplemental Figure S16.
Software availability
RCK is available at GitHub (https://github.com/raphael-group/RCK) and as Supplemental Code.
Competing interest statement
B.J.R. is a cofounder and member of the Board of Directors of Medley Genomics.
Acknowledgments
This work is supported by U.S. National Institutes of Health (NIH) grants R01HG007069 (from the National Human Genome Research Institute) and U24CA211000 (from the National Cancer Institute) and U.S. National Science Foundation (NSF) CAREER Award (CCF-1053753) to B.J.R.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.256701.119.
References
- ↵Aganezov S, Goodwin S, Sherman RM, Sedlazeck FJ, Arun G, Bhatia S, Lee I, Kirsche M, Wappel R, Kramer M, 2020. Comprehensive analysis of structural variants in breast cancer genomes using single molecule sequencing. Genome Res (this issue). 10.1101/gr.260497.119
- ↵Alekseyev MA, Pevzner PA. 2009. Breakpoint graphs and ancestral genome reconstructions. Genome Res 19: 943–957. 10.1101/gr.082784.108
- ↵Aparicio S, Caldas C. 2013. The implications of clonal genome evolution for cancer medicine. N Engl J Med 368: 842–851. 10.1056/NEJMra1204892
- ↵Avdeyev P, Jiang S, Aganezov S, Hu F, Alekseyev MA. 2016. Reconstruction of ancestral genomes in presence of gene gain and loss. J Comput Biol 23: 150–164. 10.1089/cmb.2015.0160
- ↵Baca SC, Prandi D, Lawrence MS., Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, 2013. Punctuated evolution of prostate cancer genomes. Cell 153: 666–677. 10.1016/j.cell.2013.03.021
- ↵Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, 2011. The genomic complexity of primary human prostate cancer. Nature 470: 214–220. 10.1038/nature09744
- ↵Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. 2012. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28: 423–425. 10.1093/bioinformatics/btr670
- ↵Carroll SM, DeRose ML, Gaudray P, Moore CM, Needham-Vandevanter DR, Von Hoff DD, Wahl GM. 1988. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol Cell Biol 8: 1525–1533. 10.1128/MCB.8.4.1525
- ↵Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, 2012. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30: 413–421. 10.1038/nbt.2203
- ↵Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, 2009. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681. 10.1038/nmeth.1363
- ↵Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710
- ↵Cmero M, Yuan K, Ong CS, Schröder J, Corcoran NM, Papenfuss T, Hovens CM, Markowetz F, Macintyre G. 2020. Inferring structural variant cancer cell fraction. Nat Commun 11: 730. 10.1038/s41467-020-14351-8
- ↵Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang C-Z, Pellman DS, 2020. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 52: 331–341. 10.1038/s41588-019-0576-7
- ↵Dzamba M, Ramani AK, Buczkowicz P, Jiang Y, Yu M, Hawkins C, Brudno M. 2017. Identification of complex genomic rearrangements in cancers using CouGaR. Genome Res 27: 107–117. 10.1101/gr.211201.116
- ↵Eaton J, Wang J, Schwartz R. 2018. Deconvolution and phylogeny inference of structural variations in tumor genomic samples. Bioinformatics 34: i357–i365. 10.1093/bioinformatics/bty270
- ↵Eitan R, Shamir R. 2017. Reconstructing cancer karyotypes from short read data: the half empty and half full glass. BMC Bioinformatics 18: 488. 10.1186/s12859-017-1929-9
- ↵El-Kebir M, Satas G, Raphael BJ. 2018. Inferring parsimonious migration histories for metastatic cancers. Nat Genet 50: 718–726. 10.1038/s41588-018-0106-z
- ↵Elyanow R, Wu HT, Raphael BJ. 2018. Identifying structural variants using linked-read sequencing data. Bioinformatics 34: 353–360. 10.1093/bioinformatics/btx712
- ↵English AC, Salerno WJ, Reid JG. 2014. PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics 15: 180. 10.1186/1471-2105-15-180
- ↵Fan Y, Mao R, Lv H, Xu J, Yan L, Liu Y, Shi M, Ji G, Yu Y, Bai J, 2011. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet 52: 53–59. 10.1007/s13353-010-0007-z
- ↵Fischer A, Vázquez-García I, Illingworth CJ, Mustonen V. 2014. High-definition reconstruction of clonal composition in cancer. Cell Rep 7: 1740–1752. 10.1016/j.celrep.2014.04.055
- ↵Fontana MC, Marconi G, Feenstra JDM, Fonzi E, Papayannidis C, di Rorá AGL, Padella A, Solli V, Franchini E, Ottaviani E, 2018. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia 32: 1609–1620. 10.1038/s41375-018-0035-y
- ↵Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, 2017. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45: D777–D783. 10.1093/nar/gkw1121
- ↵Garraway LA, Lander ES. 2013. Lessons from the cancer genome. Cell 153: 17–37. 10.1016/j.cell.2013.03.002
- ↵Garsed DW, Marshall OJ, Corbin VDA, Hsu A, Di Stefano L, Schröder J, Li J, Feng ZP, Kim BW, Kowarsky M, 2014. The architecture and evolution of cancer neochromosomes. Cancer Cell 26: 653–667. 10.1016/j.ccell.2014.09.010
- ↵Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K, 2020. The evolutionary history of 2,658 cancers. Nature 578: 122–128. 10.1038/s41586-019-1907-7
- ↵Greenman CD, Pleasance ED, Newman S, Yang F, Fu B, Nik-Zainal S, Jones D, Lau KW, Carter N, Edwards PAW, 2012. Estimation of rearrangement phylogeny for cancer genomes. Genome Res 22: 346–361. 10.1101/gr.118414.110
- ↵Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Högnäs G, Annala M, 2015. The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353–357. 10.1038/nature14347
- ↵Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, 2014. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res 24: 1881–1893. 10.1101/gr.180281.114
- ↵Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, 2006. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res 16: 1465–1479. 10.1101/gr.5460106
- ↵Hirsch D, Kemmerling R, Davis S, Camps J, Meltzer PS, Ried T, Gaiser T. 2013. Chromothripsis and focal copy number alterations determine poor outcome in malignant melanoma. Cancer Res 73: 1454–1460. 10.1158/0008-5472.CAN-12-0928
- ↵Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, 2017. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27: 677–685. 10.1101/gr.214007.116
- ↵Kotzig A. 1968. Moves without forbidden transitions in a graph. Matematický časopis 18: 76–80.
- ↵Layer RM, Chiang C, Quinlan AR, Hall IM. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15: R84. 10.1186/gb-2014-15-6-r84
- ↵Li Y, Zhou S, Schwartz DC, Ma J. 2016. Allele-specific quantification of structural variations in cancer genomes. Cell Syst 3: 21–34. 10.1016/j.cels.2016.05.007
- ↵Lim G, Karaskova J, Beheshti B, Vukovic B, Bayani J, Selvarajah S, Watson SK, Lam WL, Zielenska M, Squire JA. 2005. An integrated mBAND and submegabase resolution tiling set (SMRT) CGH array analysis of focal amplification, microdeletions, and ladder structures consistent with breakage-fusion-bridge cycle events in osteosarcoma. Genes Chromosomes Cancer 42: 392–403. 10.1002/gcc.20157
- ↵Ma J, Ratan A, Raney BJ, Suh BB, Miller W, Haussler D. 2008. The infinite sites model of genome evolution. Proc Natl Acad Sci 105: 14254–14261. 10.1073/pnas.0805217105
- ↵McGranahan N, Swanton C. 2015. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27: 15–26. 10.1016/j.ccell.2014.12.001
- ↵McPherson AW, Roth A, Ha G, Chauve C, Steif A, de Souza CPE, Eirew P, Bouchard-Côté A, Aparicio S, Sahinalp SC, 2017. ReMixT: clone-specific genomic structure estimation in cancer. Genome Biol 18: 140. 10.1186/s13059-017-1267-2
- ↵Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. 2010. Detecting copy number variation with mated short reads. Genome Res 20: 1613–1622. 10.1101/gr.106344.110
- ↵Myers MA, Satas G, Raphael BJ. 2019. CALDER: inferring phylogenetic trees from longitudinal tumor samples. Cell Syst 8: 514–522.e5. 10.1016/j.cels.2019.05.010
- ↵Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, Garvin T, Fang H, Gurtowski J, Hutton E, 2018. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28: 1126–1135. 10.1101/gr.231100.117
- ↵Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, 2012. The life history of 21 breast cancers. Cell 149: 994–1007. 10.1016/j.cell.2012.04.023
- ↵Oesper L, Ritz A, Aerni SJ, Drebin R, Raphael BJ. 2012. Reconstructing cancer genomes from paired-end sequencing data. BMC Bioinformatics 13 (Suppl 6): S10. 10.1186/1471-2105-13-S6-S10
- ↵Oesper L, Satas G, Raphael BJ. 2014. Quantifying tumor heterogeneity in whole-genome and whole-exome sequencing data. Bioinformatics 30: 3532–3540. 10.1093/bioinformatics/btu651
- ↵Oesper L, Dantas S, Raphael BJ. 2018. Identifying simultaneous rearrangements in cancer genomes. Bioinformatics 34: 346–352. 10.1093/bioinformatics/btx745
- ↵O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. 10.1093/nar/gkv1189
- ↵Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, 2015. Whole–genome characterization of chemoresistant ovarian cancer. Nature 521: 489–494. 10.1038/nature14410
- ↵Pevzner PA. 1995. DNA physical mapping and alternating Eulerian cycles in colored graphs. Algorithmica 13: 77–105. 10.1007/BF01188582
- ↵Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM. 2010. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res 20: 623–635. 10.1101/gr.102970.109
- ↵Rajaraman A, Ma J. 2018. Toward recovering allele-specific cancer genome graphs. J Comput Biol 25: 624–636. 10.1089/cmb.2018.0022
- ↵Raphael BJ, Dobson JR, Oesper L, Vandin F. 2014. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med 6: 5. 10.1186/gm524
- ↵Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. 10.1093/bioinformatics/bts378
- ↵Ritz A, Bashir A, Sindi S, Hsu D, Hajirasouliha I, Raphael BJ. 2014. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 30: 3458–3466. 10.1093/bioinformatics/btu714
- ↵Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC. 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15: 461–468. 10.1038/s41592-018-0001-7
- ↵Shen MM. 2013. Chromoplexy: a new category of complex rearrangements in the cancer genome. Cancer cell 23: 567–569. 10.1016/j.ccr.2013.04.025
- ↵Sieverling L, Hong C, Koser SD, Ginsbach P, Kleinheinz K, Hutter B, Braun DM, Cortés-Ciriano I, Xi R, Kabbe R, 2020. Genomic footprints of activated telomere maintenance mechanisms in cancer. Nat Commun 11: 733. 10.1038/s41467-019-13824-9
- ↵Sindi SS, Önal S, Peng LC, Wu HT, Raphael BJ. 2012. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol 13: R22. 10.1186/gb-2012-13-3-r22
- ↵Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, Salit M, West RB, Batzoglou S, Sidow A. 2017. Genome-wide reconstruction of complex structural variants using read clouds. Nat Methods 14: 915–920. 10.1038/nmeth.4366
- ↵Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. 10.1016/j.cell.2010.11.055
- ↵Stratton MR, Campbell PJ, Futreal PA. 2009. The cancer genome. Nature 458: 719–724. 10.1038/nature07943
- ↵Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. 10.1038/nature15394
- ↵Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, 2017. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543: 122–125. 10.1038/nature21356
- ↵Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, 2010. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci 107: 16910–16915. 10.1073/pnas.1009843107
- ↵Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. 2013. Cancer genome landscapes. Science 339: 1546–1558. 10.1126/science.1235122
- ↵Von Hoff DD, Needham-VanDevanter DR, Yucel J, Windle BE, Wahl GM. 1988. Amplified human MYC oncogenes localized to replicating submicroscopic circular DNA molecules. Proc Natl Acad Sci 85: 4804–4808. 10.1073/pnas.85.13.4804
- ↵Wala JA, Bandopadhayay P, Greenwald NF, O'Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, 2018. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res 28: 581–591. 10.1101/gr.221028.117
- ↵Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, 2011. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8: 652–654. 10.1038/nmeth.1628
- ↵Weinreb C, Oesper L, Raphael BJ. 2014. Open adjacencies and k-breaks: detecting simultaneous rearrangements in cancer genomes. BMC Genomics 15 (Suppl 6): S4. 10.1186/1471-2164-15-S6-S4
- ↵Zaccaria S, Raphael BJ. 2020. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. Nat Commun 11: 4330. 10.1038/s41467-020-17359-2
- ↵Zaccaria S, El-Kebir M, Klau GW, Raphael BJ. 2017. The copy-number tree mixture deconvolution problem and applications to multi-sample bulk sequencing tumor data. In Research in Computational Molecular Biology. RECOMB 2017. Lecture Notes in Computer Science (ed. Sahinalp S), Vol. 10229, pp. 318–335. Springer, Cham, Switzerland.
- ↵Zakov S, Kinsella M, Bafna V. 2013. An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc Natl Acad Sci 110: 5546–5551. 10.1073/pnas.1220977110
- ↵Zerbino DR, Ballinger T, Paten B, Hickey G, Haussler D. 2016. Representing and decomposing genomic structural variants as balanced integer flows on sequence graphs. BMC Bioinformatics 17: 400. 10.1186/s12859-016-1258-4
- ↵Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, 2016. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34: 303–311. 10.1038/nbt.3432