
The Nuclear Receptor Superfamily Has Undergone Extensive Proliferation and Diversification in Nematodes
Abstract
The nuclear receptor (NR) superfamily is the most abundant class of transcriptional regulators encoded in the Caenorhabditis elegans genome, with >200 predicted genes revealed by the screens and analysis of genomic sequence reported here. This is the largest number of NR genes yet described from a single species, although our analysis of available genomic sequence from the related nematode Caenorhabditis briggsae indicates that it also has a large number. Existing data demonstrate expression for 25% of theC. elegans NR sequences. Sequence conservation and statistical arguments suggest that the majority represent functional genes. An analysis of these genes based on the DNA-binding domain motif revealed that several NR classes conserved in both vertebrates and insects are also represented among the nematode genes, consistent with the existence of ancient NR classes shared among most, and perhaps all, metazoans. Most of the nematode NR sequences, however, are distinct from those currently known in other phyla, and reveal a previously unobserved diversity within the NR superfamily. In C. elegans, extensive proliferation and diversification of NR sequences have occurred on chromosome V, accounting for > 50% of the predicted NR genes.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos.AF083222–AF083225 and AF083251–AF083234.]
Nuclear receptors (NRs) are one of the most abundant classes of transcriptional regulators in metazoans and are involved in processes as diverse as sexual differentiation, metabolic regulation, insect metamorphosis, vertebrate limb development, and embryonic pattern formation (Gronemeyer and Laudet 1995; Kastner et al. 1995; Manglesdorf et al. 1995; Thummel 1995). The defining members of the NR superfamily were first identified biochemically as receptors for steroid and thyroid hormones, and subsequent cloning of the genes revealed that these receptors constituted a class of related proteins. The signature motif of the superfamily is a DNA–binding domain (DBD) comprised of two Cys2–Cys2 zinc coordinating modules (Rastinejad 1997). The ability of a nuclear receptor to regulate gene expression is modulated by binding of the cognate ligand to a domain positioned carboxy-terminal to the DBD. This ligand-binding domain (LBD) also participates in receptor homo- and heterodimerization, and contributes to transcriptional regulation. Although less highly conserved than the DBD, the LBDs of the classic nuclear receptors also contain regions of conserved similarity (Simons 1997).
Additional members of the NR superfamily have been identified by sequence similarities to known NRs, primarily based on the conserved DBD motif (Manglesdorf et al. 1995). The superfamily is now represented by >400 cloned sequences from >60 species. These sequences define at least 70 distinct NRs, several of which are represented by orthologs from multiple species (Gronemeyer and Laudet 1995). A number of NRs first recognized on the basis of sequence were subsequently shown to be receptors for known hormones (Arriza et al. 1987; Giguere et al. 1987;Petkovich et al. 1987; Evans 1988; Koelle et al. 1991). Efforts in many laboratories during the past decade have brought to 23 the number of NRs for which ligands or candidate activators are known (Mangelsdorf and Evans 1995; Manglesdorf et al. 1995; Forman 1997; Forman et al. 1998). The majority of NRs remain orphan receptors for which the specific ligands, if any exist, have not yet been identified.
Extensive analysis of relationships within the NR superfamily by Laudet and coworkers defined six major NR families, most of which can be further subdivided into distinct classes of orthologs and paralogs (Laudet et al. 1992; Escriva et al. 1997; Laudet 1997). Five of these six NR families contain members from both vertebrates and insects (Laudet 1997, A. Sluder, unpubl.), indicating that these families arose before the evolutionary divergence of protostomes and deuterostomes. The exception is the vertebrate steroid receptor family, members of which have thus far not been found in other phyla. In addition, the small knirps family of divergent orphan receptors (which was not included in Laudet’s analysis) is currently represented only in dipterans. Phylogenetic analysis of the six major families led to the proposal that current NR diversity arose from two distinct waves of gene duplication and divergence: one that occurred very early during the emergence of metazoans, and a second that expanded particular NR classes in the vertebrate phylogenetic lineage (Escriva et al. 1997;Laudet 1997).
Only five nematode sequences were included in the analysis described above. Using the conserved DBD zinc modules as the defining NR motif, we find >200 predicted NR genes in the genome of Caenorhabditis elegans, five–fold more than reported for any other species to date. Analysis of available genomic sequences from the related speciesCaenorhabditis briggsae indicates that it is likely to have a comparable number of NR genes. To establish a phylogenetic framework for the functional analysis of nematode NR genes, we undertook a comparative analysis of these sequences. Specifically, we addressed two questions: First, how many of the NR sequences correspond to functional genes? Second, what are the relationships of the nematode NRs to those from other organisms? Our experimental results in combination with data available in published reports and the public sequence databases demonstrate expression for 25% of the predicted C. elegans NR genes, and statistical arguments suggest that the majority of the 228C. elegans NR sequences are likely to represent expressed genes. In addition, comparative analysis of the nematode NR DBD sequences shows that both highly divergent NR sequences and phylogenetically conserved NR classes are represented in the nematode genes. Our observations are consistent with the existence of primordial metazoan NR families and later extensive duplication and diversification of NR genes in nematodes.
RESULTS
Nematode NR genes were identified by three means — polymerase chain reaction (PCR) amplification of genomic sequences using NR-specific degenerate primers, characterization of expressed sequence tags (ESTs), and analysis of genomic sequences released by the C. elegansGenome Sequencing Project. Below we first summarize our characterization of cDNA clones and the initial analysis of NR sequences revealed by the genome project. We then present the results of our expression studies and comparative analyses.
Identification of cDNA Clones for 13 C. elegans NR Genes
C. elegans sequences encoding predicted members of the nuclear receptor superfamily were isolated by PCR amplification of genomic DNA using degenerate primers corresponding to highly conserved regions of the NR DBD (see Materials and Methods). Amplification products were cloned and sequenced to identify eight candidate NR genes (Fig. 1A). The cloned PCR products were used to probe a filter grid of ordered yeast artificial chromosomes (YACs) representing ∼95% of the C. elegans genome (Coulson et al. 1995), verifying that these sequences are represented in the C. elegans genome. As each YAC represents a defined region of the genome, these hybridizations also mapped each sequence to a known physical location within the genome. nhr-4 and nhr-6were also detected in a hybridization screen described elsewhere (Sluder et al. 1997). The predicted genes and their locations are summarized in Table 1A.

DBD sequences encoded by C. elegans NR genes described in this study. Shaded amino acids are conserved in all NRs other than the nematode exceptions noted in the text.. Arrows indicate the eight zinc-coordinating cysteines (four per zinc-binding module). Asterisks denote positions of residues found to make specific DNA contacts in the existing NR DBD X-ray crystal structures (Rastinejad 1997). Underlying rectangles in the core DBD indicate the P box sequences that are primary determinants of DNA-binding specificity and the D box dimerization interface (Gronemeyer and Laudet 1995). Also indicated are the T box and A box regions that contribute to dimerization and DNA binding, respectively, by some NRs (Gronemeyer and Laudet 1995;Rastinejad 1997). Alignments were generated using the GCG Pileup program (Devereux et al. 1984). (A) NR sequences identified by degenerate PCR amplification of genomic DNA. Amino acids encoded by the original amplification products are underlined. (B) NR sequences identified from expressed sequence tags. The nhr-14DBD sequences are from the corresponding genome project ORF prediction, as the cDNA sequenced was incomplete and did not extend to the DBD region.
C. elegans NR Genes Described in this Study
The cloned PCR products were also used as probes to screen a mixed stage C. elegans cDNA library. cDNA clones were recovered for four of the eight genes (nhr-3, nhr-4,nhr-6, and nhr-18), and the complete sequence of each of these cDNAs was determined. We also completed the sequences of cDNAs representing an additional eight C. elegans nuclear receptor genes (Fig. 1B and Table 1B) that were identified as ESTs in two different surveys (McCombie et al. 1992; Waterston et al. 1992). A recent, more extensive EST project (Y. Kohara, pers. comm.) has reported ESTs for nhr-4, nhr-6, nhr-7, andnhr-21, and we completed the sequences for the nhr-7and nhr-21 ESTs. Through this combination of library screening and EST analysis, we have characterized 14 cDNA clones representing 13 different NR genes.
Two of the genes for which we report cDNA sequences, nhr-6 andnhr-24, have also been described by others — nhr-6as ceb-1 (Wilson et al. 1992) and cnr-8 (Kostrouch et al. 1995) and nhr-24 as cnr-14 (Kostrouch et al. 1995). The nhr designation has been adopted as the standard nomenclature for nuclear receptor genes (Hodgkin 1997), and we use thenhr-6 designation here. nhr-24/cnr-14 has been shown to correspond to the sex determination gene sex-1 (Carmi et al. 1998), and we use the sex-1 designation in the remainder of this work.
The cDNAs Reveal a Variety of Features in the NR Gene Structures
As of July 1, 1998, genomic sequences corresponding to all the cDNAs but that for nhr-13 had been released by the C. elegans genome project (Wilson et al. 1994; C. elegansGenome Sequencing Project, pers. comm.). The corresponding predicted open reading frames (ORFs) are listed in Table 1 and the gene structures, except for nhr-13, are diagrammed in Figure2. Although an exact match to most of thenhr-16 cDNA sequence is found on cosmid T12C9 (chromosome II), the 5′-most 138 bp of the 154 bp exon 1 (Fig. 2, note d) are not yet represented in reported genomic sequence, which includes >25 kb of sequence upstream from the matching region. Thus, the exact gene structure and definitive location of nhr-16 remain unclear. For nhr-4, nhr-15, and sex-1 the splicing patterns predicted by the genome project are confirmed by the cDNA sequences. The splicing patterns, and thus the predicted protein products, of the remaining cDNAs differ from those predicted by the genome project.
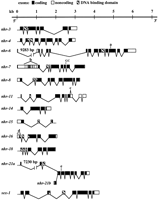
C. elegans NR gene structures defined by cDNA sequences. (Solid boxes) Coding; (open boxes) noncoding; (hatched boxes) DBD. Specific features of note (see text for additional discussion): (nhr-6) a marks site of possible alternate splice suggested by RT–PCR results. (nhr-7) vertical lines mark locations of AUG codons upstream of predicted initiation codon. Bracketb indicates possible splice sites that would remove upstream ORFs from the 5′ UTR. Also indicated is the site of a rare GC 5′ splice donor site (Blumenthal and Steward 1997). (nhr-11) bracket c marks predicted intron that would remove in-frame stop codon and extend the ORF. (nhr-16) bracket d indicates sequences without a current match in released genomic sequence. (nhr-21) e indicates the site of two splice acceptor sites separated by 6 bp; the 3′ site is used innhr-21a and the 5′ in nhr-21b.
None of the 14 cDNA clones sequenced contain the trans-spliced leader (SL) sequences found at the 5′ ends of many C. elegansmRNAs (Krause and Hirsh 1987; Blumenthal and Steward 1997), so none unambiguously pinpoints the 5′ end of the corresponding transcript. However, seven of the clones (cm2h1 and cDNAs for nhr-3,nhr-4, nhr-6, nhr-7, nhr-8, andnhr-16) have in-frame stop codons upstream of the apparent start codons and end in poly(A) tracts, indicating that these do contain complete coding regions. The EST clone CEESV11 (nhr-15) lacks the 3′ portion of the cDNA sequence, as it does not contain a poly(A) tract; the cDNA was apparently truncated at an internal XhoI restriction site during library construction. The other six clones (yk77h11 and cDNAs for nhr-11,nhr-13, nhr-14, nhr-18, and sex-1) are also unlikely to be full length, as the ORFs continue to the 5′ ends of the sequences. Although the nhr-14 cDNA sequenced does not contain sequences encoding a NR DBD motif, this cDNA corresponds to a predicted ORF (T01B10.4) that does encode a DBD.
For several of the genes, the structures provide hints of strategies that may contribute to their regulation and expression:
nhr-6
The first and fifth introns, with lengths of 9283 bp and 1815 bp, respectively, are unusually large for C. elegans (Blumenthal and Steward 1997), and the gene spans >15 kb of genomic sequence. The first five exons in our cDNA are not predicted by the genome project nor represented in a previously reported cDNA (Kostrouch et al. 1995). The large fifth intron could contain a second promoter that would allow differential expression of an alternate transcript, as has been observed for other NR genes (e.g., Talbot et al. 1993; Kozlova et al. 1998).
nhr-7
The cDNA contains an unusually large 5′ untranslated region (UTR) of 1114 nucleotides. There are 15 AUG codons and 9 small ORFs upstream of the predicted initator codon (Fig. 2). In addition, the 5′ UTR has the potential to form several stable stem–loop structures (not shown). These features could contribute to the control of NHR-7 protein expression, as both upstream ORFs and 5′ UTR secondary structures have been implicated in the regulation of translation in other systems (Kozak 1991; Damiani and Wessler 1993;Lohmer et al. 1993; Geballe and Morris 1994; Hinnebusch 1997). For example, upstream ORFs in the 5′ UTRs of the Drosophila Ultrabithorax and Antennapedia genes and the mouse retinoic acid receptor β gene confer temporal and spatial translational control to downstream protein-coding regions (Zimmer et al. 1994; Ye et al. 1997). The ORFs in the nhr-7 5′ UTR are flanked by reasonable consensus splice sites that were not used in the cDNA sequenced (Fig. 2, note b). Translation of prostaglandin synthase in chicken embryo fibroblasts is regulated at the level of mRNA splicing (Xie et al. 1991), and a similar regulated splicing event removing the ORF-laden region of the nhr-7 5′ UTR could contribute to the control of NHR-7 expression.
nhr-11
The ninth predicted intron of the corresponding genome project ORF (ZC410.1) has not been spliced out in the cDNA, and a resulting in-frame stop codon leads to a much shorter carboxy-terminal domain than that predicted for ZC410.1 (Fig. 2, note c). For one NR gene in the dog heartworm Dirofilaria immitis, alternately spliced transcripts similar in structure to both cm7a11 and ZC410.1 are found (C. Maina, unpubl.), suggesting that both isoforms could also occur fornhr-11.
nhr-18
The large predicted ORF F44C8.3 contains two NR DBD motifs. Thenhr-18 cDNA corresponds to the 5′ portion of F44C8.3, spanning one of the two DBDs and ending in a poly(A) tail. Therefore, this predicted ORF is likely to represent two genes. The distance between the 3′ end of the nhr-18 cDNA and the next predicted in-frame AUG codon is only 99 bp, suggesting that these genes may be coexpressed as an operon (Zorio et al. 1994) encoding two NRs.
nhr-21
The two cDNAs (cm2h1 and yk77h11) exhibit different splicing patterns and encode different predicted protein isoforms that differ in their amino termini. The shorter isoform (nhr-21b, defined by cm2h1) has a truncated DBD bearing only the carboxy-terminal half of the second zinc module. This isoform is unlikely to bind DNA, but may retain an ability to heterodimerize with other NRs. Production of both DNA-binding and non-DNA-binding isoforms is reminiscent of theDrosophila E75 (Segraves and Hogness 1990) and E78(Stone and Thummel 1993) orphan receptor genes. In the case ofE75, the truncated isoform exerts a dominant negative effect on its heterodimer partner DHR3, modulating the ability of DHR3 to activate target genes (White et al. 1997). The NHR-21B isoform could perform a similar regulatory function.
The C. elegans Genome Contains a Large Number of NR-Related Genes
Seven C. elegans NR genes have been described in other studies (Table 2). In addition, the predicted genes defined by the genome sequencing project (Wilson et al. 1994; C. elegans Genome Sequencing Project, pers. comm.) reveal an abundance of NR genes in the C. elegans genome, bringing the total as of July 1, 1998, to 228 predicted genes (a complete listing is available at http://www.uga.edu/∼cellbio/). As 225 of these genes are found in the 85% of the genome for which sequencing has been completed, the genome may contain as many as ∼260 NR genes. BLAST (Altschul et al. 1990) homology searches indicate that regions of the genome for which sequencing is in progress contain ⩾20 additional NR genes. Thus far only five NR genes have been correlated definitively with mutationally defined loci (daf-12, fax-1, odr-7,sex-1, and unc-55; Tables 1 and 2).
C. elegans NR Genes Described in Other Studies
As noted above, 10 of the 14 cDNAs we sequenced required splicing patterns different from the initial genome project predictions. EST sequences have been reported for 34 additional NR genes (Table 3) and reveal splicing patterns different from the predicted structures for 8 genes. These observations indicated that the computer-aided assembly of ORF predictions by the genome project (Waterston et al. 1997) may frequently err in detail. Because inappropriately assembled sequences would not yield optimal multiple sequence alignments for use in the comparative analysis described below, we reviewed all 228 predicted gene structures, focusing primarily on the DBD motif.
C. elegans NR Genes Represented by ESTs
In addition to F44C8.3 (see nhr-18 above), four of the predicted ORFs (C28D4.1, C50B6.8, F44C8.2, and T19A5.4) contain two NR DBD motifs. Three considerations suggest that these five predicted ORFs most likely each represent two gene products inappropriately fused by the gene prediction algorithm used. First, in each case the paired DBD motifs are embedded in more extensive regions of repeated similarity, suggesting that they arose from gene duplication events. Second, the occurrence of operons, in which closely linked ORFs are cotranscribed and subsequently separated by trans-splicing, is well documented inC. elegans (Zorio et al. 1994; Blumenthal and Steward 1997). And finally, existing cDNAs corresponding to only one-half of a “double” NR ORF confirms the production of single ORF mRNA for two of these loci (nhr-18 in Table 1 and nhr-44 in Table3). Therefore, in the analysis below we have treated each DBD motif as representing a separate gene product (designated, e.g., C28D4.1a for the 5′ DBD and C28D4.1b for the 3′).
A different situation is revealed by the EST clone yk271b11, which contains sequences from two tandem predicted genes (nhr-51 andnhr-52 in Table 3). These predicted genes may represent a single gene with the potential to produce multiple alternately spliced isoforms or to encode a novel NR bearing two DBDs. Elucidation of the exact gene structure will require additional analysis. However, at least two distinct DBDs are possible, and we have treated them as separate for the purposes of this analysis.
Of the remaining 172 NR genes represented only by genome project ORF predictions, 48 exhibited either incomplete or unusually long DBD motifs. For each of these, the genomic sequence was examined for candidate alternate splicing strategies that would result in a more conventional DBD. Three ORFs were truncated at the end of a cosmid, and for two of these the DBD could be completed by sequences from the neighboring clone. Revised splicing strategies were identified for 27 additional ORFs, and the modified DBD motifs were used in the comparative analysis below. (Revised splicing predictions are summarized at http://www.uga.edu/∼cellbio/, and the details have been submitted to the Genome Project for inclusion in the ACeDB database.) The remaining 18 predicted ORFs do not appear to encode complete DBD motifs. For 8 of these 18, sequences capable of encoding the missing regions of the DBD are present in predicted introns or 5′ untranslated sequences, but there are no candidate splice sites that would allow these to be spliced in frame with the remainder of the corresponding ORF. Some of these sequences encoding truncated DBDs may be pseudogenes, whereas others may encode NR-related proteins whose functions do not require DNA binding. Identification of an EST demonstrates that at least one gene incapable of encoding a complete DBD (nhr-65) is expressed as mRNA.
Many of the C. elegans NR Sequences Are Expressed as mRNA
The number of predicted C. elegans NR genes is fivefold greater than that reported from any other species—at present Homo sapiens runs a distant second with 44 NR sequences in GenBank. One potential explanation for the large number of predicted C. elegans NR genes is that many of the sequences could be nonfunctional pseudogenes predicted as ORFs by the GeneFinder program and other computer algorithms used in the genome project’s sequence analysis (Waterston et al. 1997). To assess how many of the NR sequences correspond to expressed genes, we used information available in published reports and the public databases, in addition to our own experimental results.
The EST database maintained by National Center for Biotechnology Information (NCBI) contained >70,000 C. elegans entries as of July 1, 1998, primarily from the large EST project under way at the National Institute of Genetics in Japan (Y. Kohara, pers. comm.). We searched the EST database with each of the predicted C. elegans NR genes, identifying 70 EST clones (∼0.1% of C. elegans ESTs) that correspond to 48 NR sequences. These 48 genes include 14 of the 20 for which we or others have demonstrated expression, bringing to 54 the number of NR genes known to be expressed as mRNA (Tables 1 T2 T3). However, as predicted transcriptional regulators, many NRs are likely to be expressed at relatively low levels, perhaps in only a few cells (e.g., odr-7, Sengupta et al. 1994) or during a restricted period of the life-cycle (e.g.,nhr-2, Sluder et al. 1997), and thus may be poorly represented in the cDNA libraries used in the EST surveys. Notably, of the five NR genes with known genetic functions, only one (sex-1) is represented in the EST database, suggesting that EST representation may underestimate the proportion of NR genes that are functional.
To probe further the proportion of expressed NR genes represented in the EST database, we assayed directly the expression of all known NR genes on Chromosome III, the first chromosome for which the nearly complete sequence was available (Genome Sequencing Project, pers. comm.). Of the four NR genes revealed by the genomic sequence of chromosome III (Table 4), nhr-6 is the only one for which an EST sequence has been reported. However, expression of all four genes was demonstrated by RT–PCR amplification from staged mRNA preparations (Table 4 and Fig. 3). Fornhr-9, nhr-10, and nhr-20, the amplification products obtained were of the sizes expected for transcripts spliced as predicted by the genome project, and the DNA sequences of the products corresponded to the appropriate gene sequences (see Materials and Methods). For nhr-6 the predicted size RT–PCR product was obtained from L4 and adult, whereas an ∼100-bp smaller product was amplified from embryo and L1. Both products were obtained from L2 and L3. DNA sequencing has verifed that both products were derived fromnhr-6. The differences between the two nhr-6 RT–PCR products was not resolved by the single sequence reads used to verify the products, but analysis of the genomic sequence revealed a potential alternate splice site in exon 11 (see Fig. 2, note a) that would yield a transcript 108 nucleotides smaller than the cDNA while maintaining the reading frame. Developmental Northern blot analysis has confirmed the expression of nhr-6 in all stages, although it would not have resolved a transcript size difference of 108 nucleotides (Wong 1997).
RT–PCR Expression Profiles of NR Genes on Chromosome III
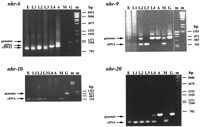
RT–PCR temporal expression profiles of NR genes on Chromosome III. RT–PCR reactions were performed as described in Materials and Methods. RNA samples were from staged cultures of embryos (E), larval stages L1–L4, and adults (A) or from mixed stage cultures (M). PCR amplification of genomic DNA (G) was included for comparison. Sizes of DNA markers (m) are indicated. Products of the sizes predicted (see Materials and Methods) from amplification of genomic DNA or cDNA samples are designated. For nhr-6, cDNA2 indicates the smaller than expected product obtained from early stages.
In summary, existing data demonstrate that at least 57 (25%) of the 228 predicted C. elegans NR genes are expressed as mRNA. Furthermore, the cDNA and EST sequences do not contain inappropriate stop codons as would appear for pseudogenes encoding nonfunctional mRNAs. Thus, mRNA expression may be a good indication that these NR sequences represent functional genes, and previously unnamed expressed genes have been assigned nhr designations (Tables 3 and 4). We will continue to refer to predicted genes for which no expression or genetic data yet exist by their genome project ORF designations.
Of the two groups of genes for which function or expression has been demonstrated by criteria other than representation in a cDNA library (genetically defined loci and the NR genes on chromosome III), only two of nine genes, or 22%, are represented in the EST database. If this proportion of EST representation holds for the NR genes in general, expression of 218 NR genes would be predicted from the existence of ESTs for 48 NR genes. We note, however, that this relatively small sample of nine NR genes may not be representative of C. elegans NR genes at large. In particular, recent progress in the sequencing of chromosomes other than III has revealed many apparent NR gene duplication events (see below), suggesting that the proportion of pseudogenes may be higher on the other chromosomes. A more comprehensive survey of NR gene expression will be needed for a definitive resolution of this issue.
NR Genes Are Also Abundant in the C. briggsae Genome
Comparisons of orthologous gene sequences between C. elegans and the sibling species C. briggsae have been used to identify conserved elements of both protein coding and regulatory regions (e.g., Kennedy et al. 1993; Wightman et al. 1993; de Bono and Hodgkin 1996; Kuwabara 1996). To allow more extensive comparisons of genome structure, the Genome Sequencing Center at Washington University has begun sequencing the C. briggsae genome. We searched the ∼5 Mb of available C. briggsae sequence (http://genome.wustl.edu/gsc/gsc/gschmpg.html) for candidate NR genes to determine whether these genes are also abundant in the C. briggsae genome. If C. briggsae also contains ∼260 NR genes, the portion of the genome sequenced (∼5%) should contain ∼13 NR genes. Fourteen likely NR genes were identified, consistent with the prediction that NR sequences are also numerous in C. briggsae.
We used two criteria to assess orthology between the C. elegans and C. briggsae NR genes. First, we required >80% amino acid identity within the DBD sequence, as most pairs ofC. elegans and C. briggsae homologs exhibit this degree of similarity (de Bono and Hodgkin 1996) and as apparent nematode/vertebrate or nematode/insect orthologous NR pairs (see below) share ⩾70% amino acid identity within the DBD. Second, we looked for synteny between the two species for other potential coding regions on the clones bearing the NRs (Kuwabara and Shah 1994). All of the NR pairs with >80% amino acid identity in the DBD also occur in genomic regions exhibiting synteny, and no NR pairs with lower degrees of similarity are in syntenic regions. By these criteria, eight of theC. briggsae NRs have orthologs in C. elegans. A complete listing of the C. briggsae NRs and, when relevant, their C. elegans orthologs is available athttp://www.uga.edu/∼cellbio/.
The Nematode NRs Exhibit Unprecedented Diversity in DBD Sequences
Several regions of NR DBDs are very highly conserved (see Fig. 1;Rastinejad 1997). Many of these conserved regions are known to be important for DNA binding or for the secondary structure of the domain (Rastinejad 1997). Whereas these elements are generally conserved in the nematode NRs, for each the sequence diversity observed in nematodes is much greater than that found in NRs currently known from other species. Among the C. elegans NRs, a number of changes are observed in amino acids that are otherwise absolutely conserved within the superfamily: replacement of one of the two adjacent conserved phenylalanine residues within the first zinc module by a leucine (F16H9.2) or a tyrosine (C17E7.5, F41B5.10); replacement of the methionine at the carboxyl boundary of the DBD by a leucine (T07C5.2, T07C5.3, T09E11.2, nhr-43); and replacement of one of the zinc-coordinating cysteines by a tyrosine (T19A5.4a). This latter change is likely to result in a nonfunctional DBD. Six of these amino acid changes could result from a single nucleotide change. To verify that these represent true diversity within the NR superfamily and not sequencing errors, each codon in question was double checked in the original sequence data by the Genome Sequencing Center, and all were found to be covered by unambiguous sequence reads (J. Spieth, pers. comm.). Whether these changes indicate that the sequences in question encode divergent NR proteins or are pseudogenes remains to be determined, although we note that at least one (nhr-43) is expressed.
Of particular interest is the sequence diversity of the P-box region (see Fig. 1). These amino acids are primary contributors to NR DNA-binding specificity (Rastinejad 1997). To date 76 distinct P box sequences have been observed within the NR superfamily. Six of these (CDGCAG, CDGCKG, CEGCKG, CEGCKS, CESCKA, CESCKG) are found in NRs from a wide variety of species, including nematodes. Seven (CAGCKG, CDGCSG, CEACKA, CEACKV, CEACYA, CEGCKA, CGSCKV) are limited to vertebrates, primarily in the steroid receptors. The remaining 63 P box sequences (complete list is available at http://www.uga.edu/∼cellbio/) have thus far been observed only in nematodes. Therefore, many nematode NRs are likely to exhibit DNA-binding specificities distinct from those defined for members of the superfamily to date. One of these novel sequences (CRACAA) is found in nearly one-third of the predictedC. elegans NR genes.
Both Phylogenetically Conserved and Novel Divergent NR Classes Are Represented in the Nematode Genes
To examine the relationships of the C. elegans and C. briggsae NRs among themselves and with NRs from other species, we carried out a comparative analysis of the nematode core DBD sequences (as defined in Rastinejad 1997), which range in length from 64 to 82 amino acids. The 18 C. elegans NRs with truncated DBDs were omitted from this analysis. A neighbor-joining tree of selected NR sequences is shown in Figure 4. Bootstrap analysis was used to identify the most stable branches, many of which were also retained in a tree generated by maximum parsimony analysis (stable branches are noted in Fig. 4). Different selections of members from the various NR classes yield similar trees, and the topology of the tree shown is generally consistent with that of a much larger tree generated with >400 DBD sequences, including all 224 nontruncated nematode DBDs (not shown). The most stable NR groupings derived in our analysis using only DBD sequences are also consistent with those defined from a combined analysis of both DBD and LBD sequences (Laudet 1997). All eight of the C. elegans/C. briggsae orthologous pairs are supported by bootstrap confidence values >95%.
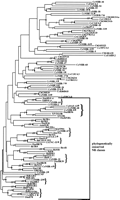
Neighbor-joining tree of selected NR core DBD sequences (as defined inRastinejad 1997) generated by the GCG Growtree program. One thousand neighbor-joining bootstrap replicates were performed on the same data set using the Paupsearch function of GCG 9.1. Bootstrap values for supported branches are indicated by hatch marks: (/) 50%-79%; (//) 80-94%; (///) 95-100%. Paupsearch was also used to perform maximum parsimony analysis; branchpoints preserved in the resulting consensus tree are denoted by dots. C. elegans sequences are included for all NR genes characterized in this or other studies (* and see Tables 1 T2 T3), all genes for which C. briggsae orthologs were identified, and selected representatives of other major groupings defined in a larger tree containing all 224 nontruncated nematode DBD sequences. Sequences not yet assigned nhr designations are indicated by genome project ORF numbers. Also included are all knownC. briggsae NR sequences (designated by clone name) and both vertebrate and insect sequences representing the major classes defined by Laudet (1997). Curly brackets indicate phylogenetically conserved NR classes that contain nematode members. Sequence name prefixes denote species of origin: (Aam) the tick Amblyomma americanum, (Cb)C. briggsae, (Ce) C. elegans, (Dm) Drosophila melanogaster, (Hs) Homo sapiens, (Xl) Xenopus laevis. Nematode sequences are available through the Genome Sequencing Project servers and, for C. elegans, in GenBank, and can be retrieved by text searches for the cosmid designation. GenBank accession numbers for previously published nematode sequences and non-nematode sequences included are (AF020187), AamEcR; (U37424), CeNHR-2; (U13075), CeNHR-23; (X51548), DmE75; (U01087), DmE78; (M74078), DmEcR; (M63711), DmFTZ-F1; (pir:S36218), DmHNF-4; (M90806), DmHR3; (X89246), DmHR38; (U31517), DmHR78; (U36792), DmHR96; (X14153,) DmKNRL; (M28863), DmSVP; (M34639), DmTLL; (X53417), DmUSP; X16155), HsCOUP-TFI; (L29496), HsAR; (X03635), HsER; (X51416), HsERR1; (U64876), HsGCNF; (U22662), HsLXRa; (M16801), HsMR; (X75918), HsNOT; (L02932), HsPPARα; (Z30972), HsPPARγ; (X06538), HsRARα; (M24857), HsRARγ; (X55066), HsReverba; (U04898), HsRORα; (U76388), HsSF-1; (Y13276), HsTL; (L27586), HsTR4; (X55005), HsTRα; (M26747), HsTRβ; (Z37526), XlHNF-4; (L11443), XlRXRγ; (X75163), XlVDLOR; (U91846), XlVDR.
Twelve of the C. elegans and 2 of the C. briggsae NRs fall into eight conserved NR classes that also contain members from other phyla (Fig. 4). In addition, the C. elegans genenhr-2 consistently groups with the conserved classes, but, as noted previously (Sluder et al. 1997), it is not a clear member of any single NR class. Nematodes are generally thought to have diverged from other metazoan phyla during the Cambrian expansion 750–650 million years ago (Fitch and Thomas 1997; Ayala et al. 1998). The conservation of particular NR classes in nematodes as well as in vertebrates and insects strongly supports the proposed ancient origin of these classes (Escriva et al. 1997; Laudet 1997). Furthermore, their continued conservation in nematodes suggests that they perform key biological functions. Consistent with this prediction, mutations in four of theC. elegans members of conserved NR classes affect key aspects of development (see Tables 1 and 2; Walthall and Plunkett 1995;Wightman et al. 1997; Antebi et al. 1998; Carmi et al. 1998).
The remaining 209 nontruncated nematode DBD sequences are divergent from all previously known NR classes. Neighbor-joining branch lengths, which are proportional to amino acid differences, are in general greater among these divergent NRs than among the conserved classes (Fig. 4), implying either a longer evolutionary history or a greater rate of evolutionary change for the divergent NRs. The latter currently seems more likely, as the divergent NRs appear to be specific to nematodes (see below) and thus to be more recently derived than the phylogenetically conserved NR classes. Detailed elucidation of the evolutionary relationships among these genes will require additional information, such as intron positions (e.g., Robertson 1998) and the relationships among their carboxy-terminal (ligand binding) domain sequences. Nevertheless, some relatively stable groups of related sequences can be defined among the divergent nematode NRs based on the DBD sequences. In general, similarities among members of a group extend beyond the core DBD through the T box but not the A box region (Fig. 1; data not shown). The T box region of the retinoid X and thyroid hormone receptors contribute to receptor dimerization (Gronemeyer and Laudet 1995; Rastinejad 1997 and references therein). However, the divergent “T boxes” differ from those of RXR and TR and their functional roles remain to be determined.
Gene Duplication and Divergence within the Nematode Evolutionary Lineage Contributed to the Abundance and Diversity of NR Genes
Most molecular screens to identify new NR genes with degenerate oligonucleotide probes (e.g., Escriva et al. 1997; Sluder et al. 1997, this work) would have been unlikely to detect many of the divergent NRs revealed by the nematode genome sequences. This raises the question of whether these divergent NRs define classes that are unique to nematodes, or represent classes not yet identified in other phyla because of the bias of the screens performed to date. Our comparative analysis of DBD sequences used all NR sequences from other species available in the main GenBank and European Molecular Biology Laboratory (EMBL) databases, all of which are members of one of the major NR subfamilies defined by Laudet (1997). The separate EST database maintained by GenBank contains >1 million sequence entries and provides a source for identification of novel gene sequences. To determine whether the EST database entries for other species contained NR sequences that would be candidate members of the NR classes represented by the divergent nematode NRs, we searched the database with 25 different C. elegans NR peptide sequences representing at least 15 different groups of the divergent NRs. When these searches were performed (April 1998), the EST database contained >900,000 entries for human, >300,000 for mouse, >70,000 for C. elegans, >30,000 for Drosophila, and smaller numbers of entries for a variety of other species. The only vertebrate NR sequences detected in these searches corresponded to genes already represented in GenBank, and are all members of previously known NR classes. One novel Drosophila NR sequence was detected; this appears to be most closely related to the vertebrate estrogen receptor-related orphan receptors (Giguere et al. 1988) and thus is also a member of a previously defined NR class. Therefore at present the divergent NR classes are found only in nematodes, although this may change as genome-sequencing projects for other species progress.
This analysis suggests that the majority of the nematode NRs may have arisen from an extraordinary proliferation of NR genes within the nematode phylogenetic lineage. Consistent with this proposal, comparisons of C. elegans and C. briggsae NRs indicate that some diversification of nematode NRs has occurred relatively recently. Molecular data suggest that many of the major nematode orders arose by 400 million years ago (Vanfleteren et al. 1994), whereas C. elegans and C. briggsae diverged between 180 and 40 million years ago (Kennedy et al. 1993), although in the absence of a nematode fossil record these evolutionary times remain uncertain. If the proliferation of NR genes occurred early within nematode evolution, the majority should be represented by orthologs in both C. elegans and C. briggsae. In this case, with ∼85% of the C. elegansgenome sequence completed, 12 of the 14 C. briggsae NRs should have orthologs among the known C. elegans genes. However, only eight (57%) of the C. briggsae NRs have clear orthologs inC. elegans. Furthermore, although the C. briggsae NR genes found in physical clusters (two clusters of two genes each) are related to specific C. elegans NR sequences, cluster structure is not conserved between the two species. On the basis of these observations, we suggest that although some of the diverged NRs were present in the common ancestor of C. elegans and C. briggsae, the process of NR proliferation and diversification also continued after the divergence of the two species.
The Genomic Distribution of C. elegans NR Genes Reveals Extensive Duplication and Diversification on Chromosome V
Over half of the predicted NR genes (145 of 228) lie on Chromosome V, whereas Chromosome III, in contrast, bears only four NR genes (Fig.5). Although V is physically the largest chromosome (∼21.7 Mb, calculated as described in Barnes et al. 1995) and III is the smallest (∼11.6 Mb), the difference in physical size is less than twofold and not sufficient to account for the difference in NR gene numbers. The density of NR genes on V (6.7 NR genes/Mb) is ∼5-fold greater than that on any other chromosome, and ∼20-fold greater than that on III (0.3 NR gene/Mb). The overall densities of ORFs predicted by the genome project, however, vary by less than twofold among the chromosomes (J. Spieth, pers. comm.). Thus, Chromosome V is enriched for predicted NR ORFs relative to the other chromosomes. The presence of pseudogenes is one possible explanation for the abundance of NR sequences on Chromosome V. Consistent with this idea, the proportion of NR sequences on V known to be expressed is lower than on the other chromosomes (14% compared to 29%–100%). However, as noted above, the current cDNA/EST representation may be an unreliable measure of the proportion of NR genes that are expressed, and a more direct survey of NR expression will be needed to address this issue fully.
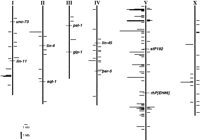
Chromosomal distribution of C. elegans NR genes. Vertical lines indicate relative physical lengths of chromosomes. Named loci designate the autosomal genetic cluster boundaries defined by Barnes et al. (1995). Histograms to the right represent NR genes with known genetic function or known to be expressed as mRNA. Those to the left represent NR sequences for which expression or genetic function has yet to be demonstrated. Physical distances were calculated as described inBarnes et al. (1995).
In addition to the large number of NR sequences on Chromosome V, a number of other observations also suggest that much of the diversification of C. elegans NRs has occurred on Chromosome V. All of the NR genes on V encode members of divergent classes, whereas the genes encoding members of phylogenetically conserved classes are distributed among the other chromosomes. Of the nine clusters of five or more tandemly arrayed NR ORFs, eight occur on V, indicating that frequent gene duplication events have contributed to the abundance of NR ORFs on V. Furthermore, two observations indicate that some of the expansion of NR sequences on V occurred after the evolutionary divergence of C. elegans and C. briggsae. First, both physical clusters of C. briggsae NRs are most closely related to C. elegans Chromosome V sequences, but, as noted above, cluster structure is not conserved between the two species. Second, only two (25%) of the eight C. elegans genes for which clear C. briggsae orthologs have been identified are on Chromosome V, compared to the 64% of all C. elegans NRs found on this chromosome. Although the current sample size is too small to be definitive, this suggests that C. elegans Chromosome V sequences may be more diverged from C. briggsae sequences than is true for the genome at large. It will be of interest to learn whether Chromosome V sequences other than NRs exhibit a similar diversity. If they do, one prediction is that as analysis of theC. briggsae genome progresses less synteny will be observed with Chromosome V than with other regions of the C. elegans genome.
DISCUSSION
The nuclear receptors, with 228 predicted genes, constitute the largest family of transcriptional regulators encoded in the C. elegans genome (Waterston et al. 1997; J. Spieth, pers. comm.). This is also the largest number of NR genes yet described from a single species, revealing a previously unobserved breadth of variation within the NR superfamily. Members of both phylogenetically conserved and novel diverged NR classes are found in the 228 genes, although the majority are divergent NRs. Although members of the new NR classes defined by the C. elegans genes have not yet been reported from other metazoan phyla, many of these divergent classes are likely to have been missed in the molecular screens for NR sequences that have been performed. Thus, these or other as yet unobserved NR classes could remain to be discovered in other metazoans, and the nature of any new NR genes revealed as other metazoan genome sequencing projects progress will be of great interest.
The most thorough analysis of the evolution of the NR superfamily to date proposed that two distinct rounds of NR gene duplication and divergence have occurred—one early in metazoan evolution, before the divergence of protostomes and deuterostomes, and a second that expanded a subset of NR classes in the vertebrate lineage (Laudet 1997). Our observations on the NR superfamily in nematodes support the hypothesis of an early presence of several ancient NR classes shared among most, and perhaps all, metazoans. In addition, our analysis indicates that extensive duplication and diversification of NR genes has occurred within the nematode lineage, resulting in an abundance of divergent NR sequences that may be unique to nematodes. Although orthologs of some divergent nematode NRs may yet be identified in other phyla, two observations are consistent with the hypothesis that at least a portion of the observed NR diversity is nematode specific. First, at least two physical clusters of divergent NR genes appear to have been expanded differentially in the genomes of C. elegans and the sibling species C. briggsae, suggesting that NR gene duplication and divergence continued after evolutionary divergence of these two species. Second, in C. elegans there is a striking concentration of divergent NRs on a single chromosome, and current data are consistent with a relatively rapid evolution of these NR sequences. A sequencing project that has begun recently for the parasitic nematodeBrugia malayi (Blaxter et al. 1996) should provide additional information regarding the extent of NR diversification within different branches of the nematode lineage.
Phylogenetic analyses of the metazoans have traditionally concluded that the nematodes, along with other pseudocoelomates, separated from other metazoans before the divergence of the protostome and deuterostome lineages, although in the absence of a fossil record for nematodes, this placement remains under some debate (see Fitch and Thomas 1997 and references therein). A recent alternate model proposes molting as a synapomorphy for placing nematodes in an “Ecdysozoa” clade along with arthropods and other protostomes (Aguinaldo et al. 1997). If molting does represent a defining shared evolutionary trait, key elements of the genetic circuitry regulating the molting process should be found in both insects and nematodes. In Drosophila melanogaster the receptor for the molting hormone 20-hydroxyecdysone is a heterodimer of two NR proteins, EcR and ultraspiracle (Yao et al. 1992, 1993; Thomas et al. 1993). Strikingly, no apparent members of the EcR or ultraspiracle NR gene classes have yet been found in C. elegans, although vertebrate genes related to both EcR (FXR; Forman et al. 1995a) and ultraspiracle (RXR; Oro et al. 1990) are known. Such genes may occur in the ∼15% of the C. elegans genome for which the sequence is yet to be completed, but no strong candidates were detected in the portions of sequence in progress available as of July 1, 1998, via the Genome Sequencing Project’s BLAST server. In addition, several direct screening strategies have failed to identifyC. elegans EcR or ultraspiracle orthologs (C. Maina, unpubl.). The absence of a C. elegans ultraspiracle/RXR ortholog, if true, is particularly puzzling as theultraspiracle/RXR class appears to be one of the more ancient NR classes (Escriva et al. 1997).
C. elegans does not produce ecdysteroids (Barker et al. 1990;Chitwood and Feldlaufer 1990), and any hormonal signal used in the regulation of molting is likely to be different from arthropod ecdysteroids. Thus if molting is a synapomorphy, the crucial conserved regulatory genes may be involved in execution of the molt rather than reception of a hormonal signal. The cellular response cascade activated by the ecdysone receptor includes additional NRs (Thummel 1995), andC. elegans genes related to several of these have been identified (Fig. 4). nhr-6, nhr-23, andnhr-25 are clear members of NR classes containing theDrosophila genes DHR38 (Kozlova et al. 1998),DHR3 (Koelle et al. 1992), and ftz-f1 (Lavorgna et al. 1991, 1993), respectively. Three C. elegans genes —daf-12, nhr-8, and nhr-48 — are most similar to the ecdysone responsive DHR96 gene (Fisk and Thummel 1995). sex-1 is the closest known C. elegansrelative of two Drosophila ecdysone-inducible genesE75 and E78, although it is not a clear ortholog of either gene (Laudet 1997). The sex-1 mutant phenotype does not reveal any role in the regulation of molting (Carmi et al. 1998), but others of these genes are candidates to participate in the molting process. Notably, disruption of nhr-23 function leads to defects in molting (Kostrouchova et al. 1998). daf-12 mutants fail to progress properly through the later larval developmental stages, instead inappropriately repeating aspects of earlier stages (Antebi et al. 1998). This daf-12 mutant phenotype is reminiscent of the reiteration of earlier stage-specific characteristics by lepidopterans exposed to exogenous juvenile hormone during larva-to-pupa or pupa-to-adult molts (for review see Riddiford 1994). Although nhr-23 and daf-12 provide intriguing parallels to the regulation of molting in insects, a full evaluation of the evolutionary significanc of these parallels will require a more complete understanding of the control of molting in nematodes.
Two additional questions of considerable interest for understanding the roles of the NR superfamily in both evolution and nematode biology are whether any of the 228 C. elegans NRs are ligand-regulated and the identities of any ligands. As with the DBDs, the C. elegans LBDs are more diverse than those yet reported from other species, and a full analysis of the nematode LBD sequences will be presented elsewhere. Preliminary analysis indicates that ∼30% of the nematode NRs, including all members of phylogenetically conserved NR classes except nhr-48, exhibit similarity to NR LBDs from other phyla, although the similarities are not sufficient to permit strong predictions regarding the identity of any ligands (A. Sluder, unpubl.). Strikingly, <10% of the divergent NR sequences on Chromosome V exhibit such LBD similarity.
The diversity of the nematode LBD sequences provides the potential for binding an equally diverse array of ligands. An organism with the small size and rapid life cycle of C. elegans seems an unlikely candidate to use enough hormones, in the classic sense, to use such a large number of receptors. One possibility is that many of the nematode NRs are not ligand-regulated, serving instead as “mere” transcriptional regulators. On the basis of currently known ligand/NR interactions, ligand binding appears to have evolved multiple independent times from a primordial NR that was not ligand regulated (Escriva et al. 1997), consistent with the view that many orphan receptors do not bind ligands.
An alternative view of NR evolution, summarized by Yamamoto (1997), is that NRs “evolved in metazoans specifically to exploit simple lipophilic molecules for intercellular signaling,” and furthermore that each NR “will bind to a small metabolite, a nutrient, an environmental compound, that has acquired a signaling role.” Consistent with this prediction, a number of vertebrate NRs are specifically activated by metabolic products with previously undiscerned signaling functions (Forman et al. 1995a,b, 1998; Kliewer et al. 1995; Janowski et al. 1996; Lehmann et al. 1997; Blumberg et al. 1998). The ability to adjust its repertoire of active metabolic pathways for optimal utilization of whatever food source is at hand would be an advantage for C. elegans in its ecological niche of opportunistic soil-dwelling bacteriovore. NR proteins are well suited to contribute to such adjustment, with the capacity to regulate specific groups of genes in response to the presence of a ligand either encountered in the environment or produced from the catabolism of a particular food source. The diversity and number of NR genes, and also of predicted chemoreceptor genes (Robertson 1998), could be major components of the genetic sophistication permitting C. elegansto exploit its environment for successful growth and reproduction. One expectation arising from this model is that nematodes adapted to different life styles, such as parasitism, will have a subset of NRs specific for their environment and not shared with C. elegans.
To date only five mutationally defined loci have been shown to correspond to NR genes (Tables 1 and 2). Several factors could be contributing to this low representation of NRs among known genetic loci. First, as noted earlier, some of the predicted NR ORFs may be pseudogenes with no genetic function. Second, as transcription factors with the potential for heterodimerization, some NRs are likely to function in multiple developmental contexts. The mutant phenotypes resulting from disruption of the genes for these will be pleiotropic and are perhaps unlikely to have attracted the attention of workers interested in specific events. Many of these genes may be represented in collections of relatively uncharacterized lethal mutants (e.g.,Hirsh and Vanderslice 1976; Meneely and Herman 1979; Cassada et al. 1981; Rogalski et al. 1982; Rosenbluth et al. 1988; Howell and Rose 1990; Johnsen and Baillie 1991; McKim et al. 1992). Third, more than one related NR may be capable of fulfilling some functions, therefore loss of a single gene may have little or no visible effect on the animal. Such genetic redundancy has been observed for the RXRs in mammals (Krezel et al. 1996). Fourth, NRs involved in the differentiation or function of a small number of cells may exhibit subtle mutant phenotypes unlikely to have been detected in many of the phenotypic screens that have been done. Notably, mutations in three of the genetic loci known to encode NRs — fax-1, odr-7, and unc-55 — each affect only a subset of neurons (Sengupta et al. 1994; Walthall and Plunkett 1995; Wightman et al. 1997). And finally, the mutant phenotypes of some NR genes, such as any involved in metabolic adaptation to environmental conditions, may not be apparent under laboratory culture conditions. Mutations in these genes may have little effect even under the majority of growth conditions in the wild, exhibiting a “latent selection potential” (Kimura 1990;Meagher 1995) that is realized only under specific circumstances.
Although they represent the largest family of predicted transcriptional regulators encoded in the C. elegans genome, the NRs are a largely untapped source of insight to nematode biology. Characterization of the expression and genetic function of these genes promises to contribute significantly to our understanding of many aspects of development and physiology. Furthermore, the diversity revealed by the C. elegans NR sequences provides an expanded context for evaluating the evolution of this family of metazoan transcription factors. In particular, identification of NRs shared among nematode species but not present in other phyla may provide a foundation for development of new strategies for combating parasitic nematode infections.
METHODS
General Methods
Unless otherwise noted, all molecular biology procedures used standard methods (Sambrook et al. 1989). DNA sequencing and oligonucleotide synthesis were carried out at the New England Biolabs core facility or the Molecular Genetics Instrumentation Facility at the University of Georgia.
Databases Used
In addition to the GenBank and dbEST databases maintained by the NCBI, the analyses reported here used the genome sequence databases of the C. elegans Genome Project. These databases are accessible for both BLAST searching and sequence retrieval through servers maintained by the Genome Sequencing Centers at Washington University, St. Louis, MO (http://genome.wustl.edu/gsc/gsc/gschmpg.html) and at the Sanger Center, Cambridge, England (http://www.sanger.ac.uk/Projects/C_elegans/). Genome sequence data for C. briggsae are also available through the Washington University server. Locations of clones on the physical map of the C. elegans genome were determined from the ACeDB database (Eeckman and Durbin 1995), data release WS4 4-26 (2/98). NR sequences were identified by BLAST homology searches (Altschul et al. 1990) of the databases using the amino acid sequence of the NHR-2 DBD (Sluder et al. 1997).
Oligonucleotide Primers
Degenerate oligonucleotide primers were designed based on the DBD sequences of the Drosophila EcR (Koelle et al. 1991) and Ultraspiracle (Oro et al. 1990) proteins or of anultraspiracle-related gene from the dog heartworm D. immitis (primers 137–126 and 141–37; C. Maina, unpubl.). Degeneracies were accomplished either by incorporation of a mixture of two nucleotides at a given position or by use of inosine to approximate four-fold degeneracy. Additional sequences added at the 5′ ends of the oligonucleotides provided restriction sites for cloning of amplified products. Sequences of the primers used were as follows (the corresponding amino acid sequences are noted in brackets): C1, 5′-ACAGAATTCTG(C/T)GA(A/G)GGITG(C/T)AA(A/G)GGITT(C/T)TT-3′ [CEGCKGFF]; C2, 5′-ACAGGATCCATICCIACIGCIA(A/G)(A/G)CA(C/T)TT(C/T)TT-3′ [reverse KKCLAVGM]; C3, 5′-ACAGGATCCT(C/T)TG(A/G)TAIC(T/G)(A/G)CA(A/G)TA(C/T)TG-3′ [reverse CQYCRYQK]; C4, 5′-ACAGAATTCGA(A/G)(C/T)TITG(C/T)TIGTITG(C/T)GGIGA-3′ [ELDLVDGC]; 137–126, 5′-ACAGGATCCCATICCCATI(G/C)(A/T)C/T)TG(A/G)CA(C/T)TTIC-3′ [reverse RKCQSMGM]; 141-37, 5′-ACAGGATCCGGIGTITA(C/T)(A/T)G/C)ITG(C/T)GA(A/G)GGITG-3′ [GVYSCEGC].
Primers for reverse transcription–polymerase chain reaction amplification were designed based on known (nhr-6) or predicted (nhr-9, nhr-10, and nhr-20) splicing patterns. nhr-6–CD-1, 5′-CCAGAGGTAGGGTCAGGGACAACG-3′; nhr-6–CD-2, 5′-GGCGCGAATGCCTGAGGCTCCC-3′; nhr-6–CD-3, 5′-CCTGGTCGACGAGAGGTTTGTTGG-3′; nhr-6–CD-4, 5′-CCTGCAACCGCCGCACCGCATTACG-3′. nhr-9–PEP9-5, 5′-CTCAACTTCTTCTGAACGCC-3′; nhr-9–PEP9-6, 5′-AAGCACAACTGCCCAATATAC-3′; nhr-9–PEP9-7, 5′-GTTCCATAAATCCCCATTCC-3′. nhr-10–PEP10-5, 5′-CGTCATCAAATAACTCGTCTC-3′; nhr-10–PEP10-6, 5′-CTTCTTTCATTTCAACAGCC-3′; nhr-10–PEP10-7, 5′-CACTCATCACTGGCTTCAAC-3′. nhr-20–PEP20-5, 5′-ATTCCACCCACATCCAATG-3′; nhr-20–PEP20-6, 5′-AATAGTTCGAAAAGTTCCGCC-3′; nhr-20–PEP20-7, 5′-TCCAGAGCATTCAAACTTGCAC-3′.
Identification of C. elegans NR Sequences by PCR Amplification Using Degenerate Primers
Candidate NR sequences were amplified from C. elegansgenomic DNA using the degenerate primers described above. Each 100-μl amplification reaction included 200–400 ng C. elegans genomic DNA; 200 μm each dATP, dCTP, dGTP, and dTTP; 1 unit Taq DNA polymerase; and 100 pmoles of each primer. After a 4-min denaturation at 94°C, reactions were cycled through 40 repetitions of 1 min at 94°C, 1 min at the selected annealing temperature, and 3 min at 72°C, followed by a final incubation at 72°C for 5 min. For each primer pair, annealing temperatures were varied in 5°C increments from 25°C to 55°C. Products that were reproducibly amplified at any specific temperature were gel purified, digested with appropriate restriction enzymes, cloned into the Litmus38 vector (New England Biolabs), and sequenced.nhr-3, nhr-4, and nhr-5 were identified using the primer pair C1/C2, nhr-6 and nhr-7 with C1/C3, nhr-17 and nhr-18 with C4/C2, andnhr-41 with 141-37/137-126.
Cloned products verified as NR sequences were used to probe filters of gridded YAC clones representing ∼95% of the C. elegans genome (obtained from A. Coulson, Sanger Center, Cambridge, England) and to screen a mixed stage C. eleganscDNA library (Stratagene). cDNAs were identified for nhr-3,nhr-4, nhr-6, and nhr-18, and the DNA sequence of each clone was determined. The GenBank accession numbers are, respectively, AF083222, AF083223, AF083224, and AF083232.
Expressed Sequence Tag Clones
NR ESTs were identified by searching the dbEST database maintained by NCBI. The corresponding cDNA clones (see Table 1) were obtained from The Institute for Genomic Research, Bethesda, MD (CEESV11 and CEESU44), the Genome Sequencing Center, Washington University, St. Louis, MO (all clones beginning with “cm”), or Y. Kohara at the National Institute of Genetics, Japan (clones beginning with “yk”). The complete DNA sequences of these clones was determined. We found that two separate samples of the clone cm06h9 did not yield sequence corresponding to that reported in dbEST or encode an NR. The cm11f5 cDNA sequence fornhr-24/sex-1 revealed no differences from a previously reported cDNA (GenBank U13074; Kostrouch et al. 1995). Accession numbers for the other completed EST sequences are AF083225 throughAF083231, AF083233, and AF083234.
Isolation of RNA from Staged Cultures
Staged cultures of the C. elegans wild-type N2 strain (Brenner 1974) were grown as previously described (Sluder et al. 1997). Total RNA was purified by LiCl precipitation (Ausubel et al. 1991) or with Tri Reagent (Molecular Research Center, Inc., Cincinnati, OH). For the latter, worms were vortexed for 10 min in Tri Reagent with a small quantity of sand (Sigma catalog S-9887) to break the cuticles. The manufacturer’s protocol for extraction of RNA from tissues was then followed. Selection of poly(A)+ RNA was as described (Sambrook et al. 1989).
RT–PCR Amplification
First strand cDNA was produced by reverse transcription of 1 μg poly(A)+ RNA with MMLV reverse transcriptase (Epicentre Technologies, Madison, WI). After phenol extraction and ethanol precipitation, cDNA products were resuspended in 10 mm Tris (pH 7.4)/1 mm EDTA. PCR amplification of 1/50 of the cDNA preparation used standard reaction conditions (Sambrook et al. 1989). After an initial denaturation step at 94°C for 2 min, 35 amplification cycles of 93°C for 1 min, 60°C for 1 min, and 72°C for 4 min were performed, followed by a final incubation at 72°C for 3 min. Secondary amplifications were performed using 1/100 of the original reaction product as template.
The following primer pairs were used for amplification. Fornhr-6, initial amplification was with CD-3 and CD-4, secondary amplification with CD-1 and CD-2; the final genomic amplification product is 1.0 kb and the predicted product from cDNA is 898 bp. Fornhr-9, initial amplification was with PEP9-5 and PEP9-6, secondary amplification with PEP9-5 and PEP9-7; product sizes are 800 bp for genomic DNA and 420 bp for cDNA. For nhr-10, initial amplification was with PEP10-5 and PEP10-6, secondary amplification with PEP10-5 and PEP10-7; product sizes are 1.0 kb for genomic DNA and 330 bp for cDNA. For nhr-20, initial amplification was with PEP20-5 and PEP20-6, secondary amplification with PEP20-5 and PEP20-7; product sizes are 600 bp for genomic DNA and 340 bp for cDNA.
Several observations support the specificity of the RT–PCR reactions. The nhr-6 cDNA was isolated and characterized at New England Biolabs, whereas the RT–PCR reactions were performed at the University of Georgia, thus minimizing the potential for contamination. As a further guard against cross-contamination of samples, the pipetters used to set up the amplification reactions were never used to pipette cloned DNAs or PCR products. Because no cDNAs have been isolated fornhr-9, nhr-10, and nhr-20, the observed RT–PCR products must have arisen from sequences present in the mRNA preparations used. In addition, the amplification of genomic DNA sequences with each primer set confirmed the reaction conditions and controlled for any cDNA contamination of reagents. And finally, each RT–PCR amplification product was gel-purified and a single DNA sequencing reaction performed for each. All RT–PCR products corresponded to the appropriate gene sequences, although the single reactions performed did not resolve the nature of the differences between the two nhr-6 products.
Sequence Analysis and Comparisons
Sequence assembly and analysis were performed using the GCG program package (Devereux et al. 1984). Prediction of candidate splice sites in both C. elegans and C. briggsae genomic sequences was aided by the GCG fitconsensus program using consensus tables derived from the data presented by Fields (1990). Synteny between C. briggsae and C. elegans genomic clones was assessed by searching C. elegans genomic sequences in GenBank with theC. briggsae sequence using the gapped alignment function of BLAST 2.0 (http://www.ncbi.nlm.nih.gov/BLAST/). Regions of similarity yielding BLAST scores >80 were considered significant. Regions were not considered syntenic unless multiple blocks of aligned similarity occurred outside the NR sequences, and for six of the eight syntenic regions thus identified the alignments extended throughout the length of the C. briggsae fosmid clone being assessed.
Neighbor-joining trees of NR DNA-binding sequences were produced using the GCG Pileup, Distances, and Growtree programs. To obtain reliable alignments of the zinc-coordinating cysteine residues, we modified the rescaled Dayhoff scoring matrix (Gribskov and Burgess 1986) of the GCG software package to weight cysteine–cysteine alignments more heavily than other matches, assigning the arbitrary high score of 45 to cysteine–cysteine matches. This modified scoring matrix was used in all analyses. Neighbor-joining bootstrap analysis (1000 replicates) and maximum parsimony analysis were performed using the PAUPsearch feature of GCG version 9.1.
Acknowledgments
We gratefully acknowledge the C. elegans Genome Sequencing Project and Yuji Kohara for making much sequence information publicly available. John Spieth was particularly helpful in matters relating to the genome sequence. Adam Antebi, Barry Honda, Pam Larsen, Barbara Meyer, Don Riddle, Bill Walthall, and Bruce Wightman generously communicated results before publication. We thank Barton Slatko, Laurie Mazzola, Jennifer Ware, and Mahul Ganatra for DNA sequencing, Marion Sibley for technical assistance in the cloning of nhr-41, Tim Lindblom for assistance with the sequencing of RT–PCR products, and Michael Weise for invaluable computer system support. A.E.S. thanks Judy Willis and Rich Meagher for many thought-provoking discussions concerning evolution and phylogeny. C.V.M. thanks Don Comb for encouragement and support and George Tzertzinis for many helpful discussions. This work was supported in part by grants to A.E.S. from the March of Dimes Birth Defects Foundation and the University of Georgia Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
NOTE ADDED IN PROOF
The WWW address for supplementary information on nematode NRs has been changed tohttp://cellmate.cb.uga.edu/cellbiology/nematode_nuclear_hormone_recepto.htm.
Footnotes
-
Present addresses: 4Biotechnology Facility, University of Hawaii at Manoa, Honolulu, Hawaii 96822 USA; 5University of Pennsylvania School of Medicine, 320 CRB, Philadelphia, Pennsylvania 19104 USA.
-
↵3 Corresponding author.
-
E-MAIL asluder{at}cb.uga.edu; FAX (706) 542-4271.
-
- Received August 17, 1998.
- Accepted December 22, 1998.
- Cold Spring Harbor Laboratory Press








