
Genomic Analysis of Caenorhabditis elegans Reveals Ancient Families of Retroviral-like Elements
Abstract
Retrotransposons are the most abundant and widespread class of eukaryotic transposable elements. The recent genome sequencing ofCaenorhabditis elegans has provided an unprecedented opportunity to analyze the evolutionary relationships among the entire complement of retrotransposons within a multicellular eukaryotic organism. In this article we report the results of an analysis of retroviral-like long terminal repeat retrotransposons in C. elegans that indicate that this class of elements may be even more abundant and divergent than previously expected. The unexpected presence, in C. elegans, of an element displaying a number of characteristics previously thought to be unique to vertebrate retroviruses suggests an ancient lineage for this important class of infectious agents.
Retroelements (e.g., endogenous retroviruses and retrotransposons) are a major component of eukaryotic genomes. For example, >50% of the maize genome is made up of retroelements (SanMiguel et al. 1996), whereas >90% of the genome in certain plants such as wheat and pine have been reported to be comprised of retroelements (Flavell 1986). In animals, the figures are also impressive. For example, at least 15% of the Drosophila (Capy et al. 1998) and 35% of the mouse and human (Yoder et al. 1997) genomes have been estimated to be comprised of retroelements. The biological importance of retroelements extends from their being major causes of mutation (Green 1988; Miki 1998) and disease (Kazazian 1998) to being significant factors in genome evolution (McDonald 1993, 1998;Britten 1996; Miller et al. 1996).
In an effort to identify factors that have shaped the evolution of this important component of contemporary genomes, our laboratory (Jordan and McDonald 1998,1999a,b) and others (SanMiguel et al. 1996; Kim et al. 1998; Marin et al. 1998) have recently initiated a genomics approach toward the study of retroelement evolution. The ongoing genome sequencing of a variety of model experimental organisms and humans is providing an unprecedented opportunity to examine the patterns of molecular variation existing among the entire complement of retrotransposons residing in genomes. Analysis of this data can provide novel insights into the tempo and mode of retroelement evolution. In this article we present a phylogenetic analysis of long terminal repeat (LTR) retrotransposons present within the genome of Caenorhabditis elegans. Our results indicate that there are no less than 12 distinct families of LTR retrotransposons represented within theC. elegans genome. These include one novel family that displays many features characteristic of complex vertebrate retroviruses such as the spumaviruses and the lentiviruses to which human immunodeficiency virus, HIV, belongs. This unexpected finding suggests that infectious vertebrate retroviruses may have a remarkably long ancestry and may have been components of ancient eukaryotic genomes.
RESULTS AND DISCUSSION
Distinct Families of LTR Retrotransposons Are Detected in C. elegans
The CLUSTALX program of Thompson et al. (1997) was used to align the amino acid sequences of all Cer proteins (see Methods).C. elegans LTR retrotransposons can be grouped into 12 families or distinct lineages based on the amino acid sequence of their reverse transcriptase (RT). We have designated ≥90% amino acid sequence identity of RT to define the individual families and have named the new families Cer2–Cer12( C . elegans retrotransposon) in keeping with the nomenclature previously used by Britten (1997). Multiple elements within a family are designated by a dash followed by an additional number. For example, we have identified a new element that is 99.9% identical at the nucleotide level to the previously described Cer1 and have designated it as Cer1-1. Interestingly, each Cerfamily consists of only one or two elements.
Structural Analysis
In all C. elegans LTR retrotransposons thus far identified, the Integrase (IN) is located 3′ to the RT domain of the element, which is characteristic of Gypsy–Ty3 group LTR retrotransposons and retroviruses. Table 1 lists other distinguishing characteristics of Cer elements including LTR lengths and percent (%) nucleotide identities between the 5′ and 3′ LTRs. LTR identity can be used to estimate the time elapsed since the element transposed (Jordan and McDonald 1998, 1999b;SanMiguel et al. 1998). With the exception of one element,Cer5-1, all elements with distinguishable LTRs displayed >99% nucleotide identity between 5′ and 3′ LTRs, indicating relatively recent transposition of the elements. Table 1 also lists the dinucleotides found at the end of the LTRs. Many LTR-retrotransposons and retroviruses universally begin and end with the dinucleotide inverted repeat (DIR) TG...CA (Dej et al. 1998). This DIR was found in nearly half of the Cer elements characterized. We also identified the direct flanking repeats that are the result of repair of the integration event. The flanking repeats of Cer elements consist of either 4 or 6 nucleotides with no apparent conservation between elements.
Cer Element Characteristics
Cer Protein Domains
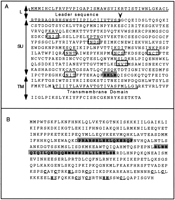
Figure 1, A–E, depicts the amino acid alignment of each protein domain from Cer elements and identifies conserved regions of each domain. The common cysteine array, Cys-X2-Cys-X4-His-X4-Cys (CCHC) of the Nucleocapsid (NC) domain of Gag (group specificantigen) was found in all elements exceptCer12 (Fig. 1A). One or two copies of the CCHC motif have been detected previously in the Gag proteins of LTR-retrotransposons and retroviruses (Coffin et al. 1997). The CCHC motif binds zinc and is thought to be important for binding to the RNA genome during element or viral assembly. Although Cer12 lacks the CCHC motif, it is unlikely that Cer12 represents a nonfunctional family of LTR retrotransposons because the homology between its LTRs (99.6%) indicates recent transposition. Interestingly, the CCHC motif has also been reported to be absent in spumaviruses and the yeast LTR retrotransposons Ty1 and Ty2 (Coffin et al. 1997). Three imperfect CCHC motifs are present in the Cer7–Cer10,Cer10-1, and Cer11 elements. The presence of three imperfect copies of the CCHC motif is also a feature of several non-LTR retrotransposons including Jockey, Doc, Het-A, and Tart-B1 ofDrosophila (Capy et al. 1997).


Cer protein alignments. The RT (C) of eachCer was identified according to the motifs previously designated by Xiong and Eickbush (1990). The additional protein domains (A = Gag, B = PR, D = Rnase H, andE = IN) were identified in a like manner using domains previously established by McClure (1991). The sequences were aligned using CLUSTAL X as described in Methods. Residue coloring was performed by MacBoxshade v2.01 and is based on the similarity scheme (F,W,Y), (I,L,M,V), (P), (D,E), (G,A), (S,T,C), (N,H),(R,K), (Q). Members of a similar residue group are the same color. In-frame termination codons or frameshifts are depicted by an X.
The Protease domain (PR) is located downstream of the NC domain in LTR-retrotransposons and retroviruses. PR is synthesized as part of the Gag-Pro-Pol precursor, usually as the result of a frameshift or readthrough (termination) suppression after Gag protein synthesis. The amino acids between the NC and RT motifs of the Cer elements are homologous to previously identified retroviral proteases. In particular, the conserved “D,T/S,G” tripeptide of retroviral PR is present in many Cer elements (Fig. 1B) (Doolittle et al. 1989).
The Pol region of retrotransposons is made up of three enzymatic domains that are processed from the Pol precursor. Pol consists of the RT, RNase H, and IN domains. An alignment of the RT domain ofCer elements is shown in Figure 1C. The characteristic motifs of RT, as previously defined by Xiong and Eickbush (1988), are present in all Cer elements (Fig. 1C). Cer elements 7, 8, 9, 10, 10-1, 11, and 12 (Cer7–Cer12) contain an unusual “YVDN” tetrapeptide motif at the active site of RT. The RNase H domains of the Cer elements were aligned, and previously identified motifs (McClure 1991) were boxed and labeled (Fig. 1D). Finally, the previously identified HHCC motif and DDE domain (Capy et al. 1996) of IN are present in all Cer elements (Fig. 1E).
Position of Cer Elements in Existing RT PhylogenyWithin Gypsy–Ty3 Group Clade
Because the RT domain has the slowest relative rate of change among all retroelement proteins (McClure et al. 1988), RT multiple sequence alignments (Xiong and Eickbush 1988; Doolittle et al. 1989) have been used to elucidate the evolutionary relationships among retroelements. Following characterization of the RT domain of the Cerelements, we made alignments with previously reported RT sequences (Fig. 2A). The most conserved regions of RT are shown boxed in Figure 2A. These regions represent the RT ordered series of motifs (OSM) (Hudak and McClure 1999). The sequences comprising the RT OSM, as well as the full-length RT domain, were used to construct neighbor-joining (NJ) phylograms (Fig. 2B) from which the phylogenetic relationships of theCer elements to other LTR-retrotransposons and retroviruses could be deduced.

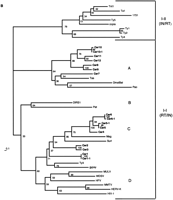
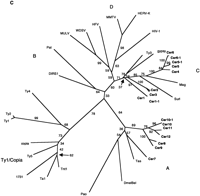
RT phylogenetic analysis. (A) RT amino acid alignment. The domains are boxed and numbered as by Xiong and Eickbush (1988). Additional RT sequences used in the Figure are referenced in Xiong and Eickbush (1990) or described as follows: (HFV) Human foamy virus, accession no. Y07725; (WDSV) walleye dermal sarcoma virus, accession no. AF033822; (PAT) Panagrellus redivivus retrotransposon, accession no. X60774; (Tas) A. lumbricoides retrotransposon, accession no. Z29712. Tas contains an in-frame termination codon at position 36 above. A gap was inserted at this position for the phylogenetic analysis. Residue coloring is based on the scheme described in Fig. 1. (B) NJ phylogram of sequences aligned inA (see methods). The tree was rooted with all of theCopia–Ty1 group LTR retrotransposons. Major clades are labeled according to the nomenclature proposed in Table 2. (C) Unrooted NJ phylogram of sequences aligned in A. Major clades are labeled as in B.
The first group, to which Cer1 belongs, clusters most closely to the Gypsy–Ty3 group of LTR retrotransposons. Other elements clustering within the Gypsy–Ty3 clade areCer2 and Cer3. These two elements group closest to the previously identified SURL element from echinoderms.Cer4, Cer5, Cer5-1, Cer6, and Cer6-1 are most closely related to the previously identified retrotransposon, Mag, from Bombyx mori and like Mag, have relatively short LTRs (see Table 1).
Outside of Gypsy–Ty3 Group Clade
Cer7–Cer12 represents a unique branch from both theGypsy–Ty3 and the retroviral groups of retroelements. This branch of Cer elements is most closely related to previously described elements Tas, Pao, and Bel isolated fromAscaris lumbricoides, B. mori, and Drosophila melanogaster, respectively (Xiong et al. 1993; Felder et al. 1994;Davis and Judd 1995). As noted above, one distinguishing characteristic of Cer7–Cer12 is the presence of the conserved tetrapeptide YVDN at the active site of the RT enzyme. This motif was previously found in the RT region of the Tas element of A. lumbricoides(Felder et al. 1994). This motif differs from the tetrapeptide “F/YXDD” found in all previously reported RT sequences. Because the dipeptide “DD” had previously been thought to be essential for RT activity, it was speculated as to whether “YVDN” was indeed functional (Felder et al. 1994). Our finding that the “YVDN” motif is present in six distinct lineages of Cer elements argues strongly for its functionality. Additionally, all calculated LTR/LTR identities for Cer7–Cer12 are >99%, indicating that they have recently transposed.
One member of this group of elements, Cer7, contains a large ORF immediately following the IN domain that displays many features characteristic of the envelope (env) encoding region of retroviruses. The size and location of this ORF is nearly identical to that of the env encoding region of theAcaris Tas retroelement (Felder et al. 1994). The presence of putative C. elegans splice donor and splice acceptor sites inCer7 indicates that a smaller (subgenomic) RNA could be formed between the 5′ leader portion of the full-length (genomic) transcript and an acceptor site located upstream of the envinitiation codon (Fig. 3). Although no significant sequence homology was found between the putative Env of Cer7and other retroviral Env proteins, this is to be expected given the nature of envelope function and the large divergence between host organisms. There are, however, many structural similarities between the putative Env of Cer7 and other retroviral Env proteins. The putative Cer7 Env contains a leader (L) signal peptide domain, multiple N-glycosylation sites, and a transmembrane region followed by a basic anchor (see Figs. 3 and 4A). Also present is a minimal furin-protease consensus cleavage site, “RXXR” (Molloy et al. 1992) (Figs. 3 and 4A), which may serve as the cleavage site between the integral surface glycoprotein (SU) and the transmembrane domain (TM). These are structural features common to all retrovirus envelope proteins (Coffin et al. 1997).
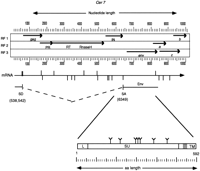
Cer7 ORF map and putative Env. The largest ORFs (>200 amino acids) from the three forward reading frames (RF1, RF2, and RF3) ofCer7 are translated and depicted as solid lines. Stop codons are shown as arrowheads. The common retroviral domains (PR and IN) and ORFS (gag and env) are labeled below the indicated ORFs. Note the three ORFs (a, b, and c) following the putativeenv ORF. Accessory proteins of complex retroviruses are found in analogous regions. The genomic mRNA is shown as a horizontal arrow below the ORF map. The predicted splice donor (SD) and splice acceptor (SA) sites are shown as vertical lines above and below the mRNA arrow, respectively. The predicted splice donor and acceptor sites that lead to the Env ORF production are shown below the mRNA. The Env protein is depicted as a box below the predicted spliced RNA. The leader peptide is indicated by an L. The possible N-glycosylation sites are depicted by a “Y” above the Env box. Vertical lines within the box represent putative protease cleavage sites that may serve to remove the leader peptide and cleave the region between the integral surface (SU) and transmembrane (TM) domains of Env. The predicted membrane anchor of TM is shown in grey. See Methods for a description of the functional motif predictions.
Amino acid sequences of putative Cer7 Env protein (A) and accessory protein C (B). (A) The leader sequence and transmembrane domain of Env are bracketed beneath and labeled. The predicted signal peptide cleavage site is indicated by a vertical arrowhead. The seven N-glycosylation targets “NXT/S” are boxed. The grey box indicates a putative furin cleavage site. Vertical double headed arrows at left of the sequence delineate the leader (L), surface (SU), and transmembrane (TM) domains of Env. (B) Gray boxes indicate the two predicted coiled-coil regions of ORF C. The mulitple cysteine and histidine residues are shown in bold and underlined. See Methods for a description of the functional motif predictions.
Another interesting feature of Cer7 is that it has three small overlapping ORFs following its putative env gene (Fig. 3). This is a feature of the complex vertebrate retroviruses such as the human T-cell leukemia–bovine leukemia viral group (HTLV, BLV), the spumavirus, and the lentivirus groups. One of the small ORFs following the putative envelope of Cer7, which we call ORF C, has a strongly predicted C. elegans splice acceptor site (D2 = 13.77) located upstream of its putative initiation codon. The predicted ORF C contains a region rich in cysteines and histidines, as well as two coiled–coil regions (Fig. 4B) that may function in DNA-binding or protein dimerization formation. These features are strikingly similar to other transactivating accessory proteins found in complex vertebrate retroviruses. For example, the HTLV-1Tax protein also contains a region rich in cysteines and histidines. Tax is thought to interact with bZip domains of ATF/CREB proteins to stably recruit CBP and other general transcription factors to the U3 region of the HTLV-1 LTR to remodel nucleosomes and activate transcription (Bex and Gaynor 1998). Likewise, spumaviruses encode proteins between their env and 3′ LTR (Bel-1 in human foamy virus and Taf in simian foamy virus) that stimulate transcription by interacting with multiple sites within the U3 regions of their LTRs. Although Tax, Bel1, and Taf have analogous functions, they are not homologous in sequence suggesting that these complex retrovirus functions may have arisen multiple times during evolution.
The presence of the retroviral-like Cer7 within the C. elegans genome indicates that endogenous retroviruses may be quite ancient components of animal genomes. It has been previously hypothesized that retroviruses originated about the time of the mammals (Temin 1985; Doolittle et al. 1989). However, our findings suggest that the ancestor of vertebrate retroviruses may have had an early metazoan origin.
Retroelement phylogeny
In general, our results indicate that LTR-retrotransposons and retroviruses may be much more abundant and divergent than previously thought. The high level of sequence divergence among the C. elegans elements and previously characterized retroelement RTs indicates that a revision of LTR-retrotransposon and retrovirus phylogeny may be in order. An unrooted phylogenetic analysis of the sequences from Figure 2A places the clade containingCer7–Cer12 between the Copia–Ty1 andGypsy–Ty3 groups (Fig. 2C). However, since theCer7–Cer12 clade is structurally similar to theGypsy–Ty3 group regarding the location of itsintegrase gene, it should be placed into theGypsy–Ty3 group. This creates a Gypsy/Ty3 group with four major branches (Fig. 2B,C). The branches leading to these four major subgroupings as well as the branch leading to theTy1/Copia group are supported by a majority of bootstrap replicates. These groupings/subgroupings include (1) the Ty1/Copia group, (2) the branch leading to the DIRS-1 and Pat elements, (3) the branch leading to the Cer7–Cer12 elements as well asPao, Bel, and Tas, (4) the branch leading to the group containing Ty3 and gypsy, and finally, (5) the branch leading to the vertebrate retroviruses. Table2 depicts a proposed nomenclature for the characterization of LTR-retrotransposons and retroviruses. Class I elements (i.e., those transposing via an RNA intermediate) can be divided first into three groups (I–III) based on defining structural differences. We propose group I to consist of LTR-retrotransposons and retroviruses with the RT/Rnase H/IN order of the Gypsy–Ty3group. Group II consists of LTR retrotransposons with the IN/RT/Rnase H order of the Copia–Ty1 group, whereas group III consists of non-LTR retrotransposons. We propose that the next level of classification in group I be based on the major branches of the RT phylogeny. This level of classification is the least exact and is likely to undergo many additions and reclassifications as additional LTR retrotransposons are described that better delineate RT relationships. Based on this criterion, there are presently four major subgroups of group I. We designate these A–D as shown in Table 2. Subgroup A contains Pao, Tas, Bel, and Cer7–Cer12. Subgroup B contains the unusual retroelements DIRS-1 fromDictyostelium discoideum that has inverted rather than direct terminal repeats and PAT from the nematode Panagrellus redivivus that has split direct repeats (de Chastonay et al. 1992). Subgroup C contains the well-known Gypsy–Ty3 group, as well as Mag from B. mori. Subgroup D contains the vertebrate retroviruses. We propose that the next level of classification be based primarily on RT amino acid pairwise identity. An RT amino acid identity of ≥50% groups an element into the level of genus as used in the retroviral nomenclature (e.g., Lenti, Spuma, Type D, etc.). A ≥75% identity groups an element into a superfamily. Finally, a ≥90% identity groups an element into a family. Family members are designated by a dash followed by a number given upon order of identification. Names of genus, superfamily, and family are designated by the names of the first member of each group as indicated in Table 2.
Class I TE Nomenclature
The genome sequencing of model experimental organisms and humans is providing an unprecedented opportunity to examine the evolutionary relationships that exist among retroelements including LTR-retrotransposons and retroviruses. Analysis of LTR retrotransposons in the C. elegans genome indicates that this group of retroelements may be more abundant and divergent than previously suspected. In addition, the presence in C. elegans of elements displaying a number of characteristics previously thought to be unique to vertebrate retroviruses suggests an ancient lineage for this important class of infectious agents.
METHODS
Sequence Identification and Retrieval
The C. elegans genomic sequence data used in our analysis was accessed directly from the web sites of the Sanger Center, Hinxton Hall, Cambridge, UK, and the Genome Sequencing Center at the Washington University School of Medicine, St. Louis, Missouri Sequence retrieval was initiated by performing TBLASTN searches against the C. elegans genomic sequence (http://www.sanger.ac.uk/Projects/C_elegans/blast server.shtml) using the RT from the sole LTR retrotransposon representative from the C. elegans genome,Cer1 (accession no. U15406) (Britten 1997). We also queried the current WORMPEP (http://www.sanger.ac.uk/Projects/C_elegans/wormpep) database for ORFs with predicted homology to RT. Additional BLAST and TBLASTN searches were performed with the putative RTs until overlapping hits were retrieved by all sequences. A number of RTs belonging to the non-LTR containing retrotransposons were also detected within the C. elegans genome. These have been described previously by Marin et al. (1998).
Complete clones were retrieved from GenBank and characterized using SeqLab: The Graphical User Interface to the Wisconsin Package, (GCG 1999) maintained and made accessible by the Research Computing Resource (RCR) at the University of Georgia (UGA) (http://www.rcr.uga.edu/ biosci/home.html). Many elements were found to span two adjoining cosmids and were first aligned and made contiguous using GAP and BESTFIT. The dot matrix program COMPARE was used to identify regions of identity within each C. elegans cosmid. DOTPLOT was used to visualize the dot matrixes generated with COMPARE. LTRs appeared as two lines parallel to the identity diagonal (one above and one below). Subsequent analysis revealed the phylogenetically conserved TG...CA dinucleotide at the ends of many LTRs. Clone sequence, position, and ORFs were also viewed in A Caenorhabditis elegans Database (ACEDB) (Durbin and Thierry Mieg 1991) (documentation, code, and data available from anonymous FTP servers at lirmm.lirmm.fr,cele.mrc-lmb.cam.ac.uk, and ncbi.nlm.nih.gov).
Multiple Sequence Alignments
The CLUSTAL X program (Thompson et al. 1997) was used to align the amino acid sequences of all Cer proteins and domains presented in Figure 1. RT amino acid sequences presented in Figure 2A were also aligned using the CLUSTAL X program. CLUSTAL X was run using the Gonnet 250 pairwise aligment and Gonnet series multiple alignment parameter settings. The Gonnet 250 and Gonnet series settings were able to recognize and align parts of all of the conserved regions of RT. These regions have been described previously (Xiong and Eickbush 1990;McClure 1993) and are collectively known as the RT OSM as described byHudak and McClure (1999). The regions between the motifs are less conserved and represent hypervariable regions of the RT domain. The larger gaps introduced by CLUSTAL X were in these hypervariable regions. Using SeqLab, manual adjustments were made around regions containing gaps to minimize mutation events and to agree with previously published multiple alignments of RT (Xiong and Eickbush 1990; McClure 1991, 1992, 1993). Three different multiple sequence alignments were used in subsequent phylogenetic analyses. One analysis consisted of the entire RT, whereas a second consisted of only residues of the RT OSM (boxed in Fig. 2A). This served to eliminate the hypervariable and ambiguous regions of the alignments. Finally, a third alignment consisted of the regions between the RT OSM. These regions are also known as motif-intervening regions (MIRs).
Phylogenetic Analysis
Phylogenetic analyses were performed on the multiple sequence alignments using distance methods used by CLUSTAL X and PHYLIP (Felsenstein 1993). Draw N-J Tree and Bootstrap N-J commands of CLUSTAL X were used to generate nonbootstrapped and bootstrapped trees, respectively. The PRODIST program of PHYLIP using the Categories model was used to generate distance matrices that were analyzed with the NEIGHBOR program to generate NJ tree files. SEQBOOT was also used to generate 100 data replicates that which were subsequently analyzed with PRODIST (Categories model), followed by NEIGHBOR, and finally with CONSENSE to generate an unrooted bootstrapped tree. Analyses using the RT OSM, the MIRs, or the entire RT domain converged on an unrooted tree with the same general topology. This indicates a strong signal-to-noise ratio with the full-length multiple sequence alignment. We report the trees and bootstrap values generated with the PHYLIP package using the entire RT domain. The additional information included in the MIRs serves to strengthen the accuracy of the within subgroup phylogenetic reconstructions. The phylogram presented in Figure 2B was rooted with the Ty1/Copia group. All trees generated were visualized with TreeViewPPC version 1.5.3, (Page 1996).
Cer7 Protein Predictions
C. elegans splice sites were predicted by the SPL program of Baylor College of Medicine's GeneFinder (Solovyev et al. 1994). The leader peptide of the Cer7 Env was predicted by SignalP version 1.1 (Nielsen et al. 1997). N-glycosylation sites were located by PROSITE (Hofmann et al. 1999). The transmembrane domain was identified by PHDhtm (Rost et al. 1995). Finally, the coiled-coil regions of Cer7 C were predicted by COILS (Lupas et al. 1991).
RT Pairwise Identities
Pairwise amino acid identity spanning the entire length of the RT sequence shown in Figure 2A was used for the Cer family characterizations. PAUP 4.0b2a (Swofford 1999) was used to calculate pairwise mean differences that were converted to percent identities.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL mcgene{at}arches.uga.edu; FAX (706) 542-3910.
-
- Received May 12, 1999.
- Accepted August 12, 1999.
- Cold Spring Harbor Laboratory Press








