
AAA+: A Class of Chaperone-Like ATPases Associated with the Assembly, Operation, and Disassembly of Protein Complexes
Abstract
Using a combination of computer methods for iterative database searches and multiple sequence alignment, we show that protein sequences related to the AAA family of ATPases are far more prevalent than reported previously. Among these are regulatory components of Lon and Clp proteases, proteins involved in DNA replication, recombination, and restriction (including subunits of the origin recognition complex, replication factor C proteins, MCM DNA-licensing factors and the bacterial DnaA, RuvB, and McrB proteins), prokaryotic NtrC-related transcription regulators, the Bacillus sporulation protein SpoVJ, Mg2+, and Co2+ chelatases, theHalobacterium GvpN gas vesicle synthesis protein, dynein motor proteins, TorsinA, and Rubisco activase. Alignment of these sequences, in light of the structures of the clamp loader δ′ subunit ofEscherichia coli DNA polymerase III and the hexamerization component of N-ethylmaleimide-sensitive fusion protein, provides structural and mechanistic insights into these proteins, collectively designated the AAA+ class. Whole-genome analysis indicates that this class is ancient and has undergone considerable functional divergence prior to the emergence of the major divisions of life. These proteins often perform chaperone-like functions that assist in the assembly, operation, or disassembly of protein complexes. The hexameric architecture often associated with this class can provide a hole through which DNA or RNA can be thread; this may be important for assembly or remodeling of DNA–protein complexes.
Nearly every major process in a cell is carried out by macromolecular machines—protein complexes with highly coordinated moving parts driven by energy-dependent conformational changes (Alberts 1998). Examples of such structures include proteasomes, spliceosomes, ribosomes, peroxisomes, and chromosomal replicases. Hence, to understand cellular processes it is important to characterize the elemental components of these machines and to find general principles associated with their assembly and function (Alberts 1998).
The intricacy of these machines is underscored by the need for additional devices to assist in their assembly. Eukaryotic chromosomal replicases, for instance, require a clamp-loader complex to load PCNA sliding clamps onto DNA (for reviews, see Stillman 1994; Kelman and O’Donnell 1995; Baker and Bell 1998). This is accomplished by coupling binding and hydrolysis of ATP to conformational changes in the clamp-loader leading to substrate remodeling and DNA binding of the clamp protein. Proteins that induce formation of a DNA–protein complex in this way have been described as molecular matchmakers (Sancar and Hearst 1993). In general, however, a role as molecular matchmaker need not be limited to DNA-binding complexes, but may involve the assembly and function of other protein complexes as well.
Such roles are usually associated with molecular chaperones—proteins that assist in the noncovalent assembly of other proteins or protein complexes. Chaperones often work together with proteases to degrade misfolded and mistranslated proteins (Horwich 1995; Gottesman et al. 1997; Suzuki et al. 1997), in which case the chaperone’s remodeling activity makes the substrate protein more accessible to proteolysis. This can provide a quality control mechanism to rid the cell of malfunctioning components that fail to integrate properly. Chaperones can also regulate the activities of protein complexes by mediating the degradation or availability of specific components.
Evolutionarily related chaperones that function in the assembly or regulation of molecular machines are likely to be associated with diverse cellular activities. Such is the case for members of the AAA family (Confalonieri and Duguet 1995; Swaffield et al. 1995; Patel and Latterich 1998), which stands for ATPasesassociated with a variety of cellularactivities (Kunau et al. 1993). AAA modules function as regulatory subunits of the eukaryotic 26S proteasome—a complex that catalyses the ATP-dependent degradation of ubiquitinated proteins (Baumeister and Lupas 1997; Baumeister et al. 1998). AAA modules also prime the assembly of various membrane-targeting protein complexes during membrane fusion (Rowe and Balch 1997). For example,N-ethylmaleimide-sensitive fusion (NSF) protein is an ATPase that, in conjunction with α-SNAP, is required for homotypic vesicle fusion (Hay and Scheller 1997; Weber et al. 1998 and references therein). NSF performs a chaperone-like function to dissociate otherwise stable complexes of vesicle and target membrane SNAP receptors (SNAREs) after one round of fusion to facilitate the next round. Other activities associated with AAA modules include peroxisome biogenesis, the assembly of mitochondrial membrane proteins, cell-cycle control, mitotic spindle formation, cytoskeletal interactions, vesicle secretion, signal transduction, and transcription (Confalonieri and Duguet 1995; Beyer 1997; Waterham and Cregg 1997; Subramani 1998).
Here, using a combination of iterative database search and multiple sequence alignment methods, we show that other chaperone and chaperone-like protein families, including DNA–protein complex “molecular matchmakers,” are also related to the AAA family. This sequence superset is designated the AAA+ class. Multiple sequence analysis suggests that these proteins share distinct structural and mechanistic features that distinguish them from other NTPases. Recently available structures for this class confirm these relationships and provide structural cognates for the sequence similarities.
RESULTS
Starting with a set of sequences related to replication factor C (RFC) proteins described by Guenther et al. (1997), PROBE (Neuwald et al. 1997), PSI-BLAST (Altschul et al. 1997), and other procedures (see Methods) were used to detect and align members of the AAA+ class. Figure 1 shows an alignment of a representative subset of >1000 of these proteins detected in the NCBI nonredundant (NR) database. It is important to stress that even though the alignment of certain motifs for some of the sequences is uncertain because of occasional divergence, for the sequence set as a whole both the aligned regions and the highlighted patterns are clearly significant. This is because the alignment procedure relies on statistical criteria (Neuwald et al. 1997) to ensure that only those regions corresponding to clearly conserved patterns are identified and aligned without manual adjustment. As a result, several previously undetected regions of subtle, yet clearly significant sequence conservation were revealed, implying an unexpected structural and functional relationship between these protein families.


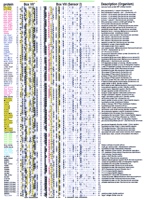
Representative multiple sequence alignment of AAA+ proteins. The NSF (NSF_CRIGR, residues 485–742) and Pol III δ′ (HOLB_ECOLI) sequences, whose structures are known, are given at the top. Sequences are grouped into families by the following color scheme: (Red) RFC and related proteins; (blue) MCMs, ORC, and Cdc6 proteins; (brown) Lon and Clp family; (teal) McrB-related proteins; (magenta) NtrC family; (yellow) AAA family; (green) dynein; (cyan) Mg2+and Co2+ chelatases; (gray) SV40 large T-antigen helicase; (black) others. Segments in individual sequences that are no more conserved than expected by chance are italicized. For each aligned column, conserved residues [i.e., elevated with binomial tail probabilities ≤ 0.01 (Neuwald and Green 1994)] and related, marginally conserved residues (with tail probabilities ≤ 0.05) are indicated using the following automated hierarchical scheme: (red highlight) ⩾2.5 bits of information; (magenta highlight) 1.8–2.5 bits of information; (yellow highlight) ⩾70% hydrophobic; (black highlight) >66% conserved; (dark gray highlight) 50%–66% conserved; (black) 33%–50% conserved; (dark gray) <33% conserved; (light gray) unconserved. Information is defined as the relative entropy of observed to background residue frequencies. The numbers of observed residues used in the relative entropy calculation were weighted by the method of Henikoff and Henikoff (1994). Related residues are defined by positive blosum62 pairwise scores (Henikoff and Henikoff 1992). Colored bars at the top correspond to the color scheme used in Fig. 2. Structural predictions are shown below the alignment. (h) Helix; (s) strand.
These structural and functional features extend well beyond the common P-loop-type NTP-binding site suggested by the Walker A and B motifs (Walker et al. 1982; Gorbalenya and Koonin 1989; Saraste et al. 1990). P-loop-type NTPases share a conserved α,β-fold core structure (Hubbard et al. 1997) and are likely to have a monophyletic origin, as indicated by the nearly identical positions of the Walker motifs in proteins of known structure. These NTPases can be classified into several major groups based on further signature motifs and clustering (Gorbalenya and Koonin 1989; Koonin 1993b; L. Aravind, unpubl.). Some of these major groups are (with characteristic signatures that can be used as a shorthand for their identification): GTPases (NKXD signature); ABC ATPases ([TS]GG signature between Walker A and B); RecA superclass group 1 (RecA-like ATPases with a GGG motif upstream of Walker A); RecA superclass groups 2 (superfamily I helicases) and 3 (superfamily II helicases), both of which typically possess a conserved α,β-fold domain carboxy-terminal to the RecA-like domain; and motif C (or sensor 1)-containing ATPases, which include superfamily III helicases (Koonin 1993a). The AAA+ class falls within this last motif C-containing group.
In addition to the Walker and motif C signatures, the AAA+ class shares other conserved regions that correspond to previously noted motifs (described as RFC boxes) shared by RFC-related proteins (Cullmann et al. 1995; Guenther et al. 1997). Although several of these conserved regions correspond to distinctive patterns located between the Walker A and motif C signatures, what distinguishes this class most clearly from other P-loop-type ATPases are several motifs beyond the motif C signature as well as an amino-terminal RFC boxII motif.
The protein families sharing these RFC box motifs are represented in Figure 1. These include Clp (Schirmer et al. 1996; Wawrzynow et al. 1996) and Lon (Gottesman 1996; Suzuki et al. 1997) protease-associated chaperones, RuvB and dynein motor proteins, and NTPases involved in DNA replication, transcription, recombination, and restriction. Regarding the Clp family, note that our analysis contradicts the reported occurrence of PDZ-like domains in the carboxy-terminal region of ClpX (Levchenko et al. 1997), which contains an AAA+ module. Furthermore, we failed to find any significant similarity in this region to a multiple alignment profile of known PDZ domains, and the structural features of this region inferred from the δ′ subunit and NSF–D2 structures are totally inconsistent with the PDZ fold (data not shown). Nevertheless, a substrate recognition role for the carboxy-terminal region of ClpX (Levchenko et al. 1997) is not inconsistent with our analysis.
Within these families, AAA+ modules occur either singly or as repeats. Notably, the huge dynein heavy chain subunit contains six modules (Fig. 1), although two of these are hard to detect because they are poorly conserved and their P-loops are disrupted. Nevertheless, these disrupted components are clearly detected within the dynein sequence by an AAA+ alignment profile (P < 0.01). Moreover, a PSI-BLAST search of the entire database using one of these poorly conserved regions detects significant similarity to a yeast hypothetical protein that also contains six AAA+ modules (P = 0.00001). Thus, these dynein AAA+ modules may form a hexameric-like assemblage—a possibility that, to our knowledge, has not been suggested previously. Interestingly, one of these AAA+modules bears a mutation in the axonemal dynein that results in the situs inversus phenotype in mice (Supp et al. 1997).
Structural Features of the AAA+ Class
The recently determined structures of the δ′ subunit ofEscherichia coli DNA polymerase III (Pol III) (Guenther et al. 1997) and of the NSF-D2 hexamer (Lenzen et al. 1998; Yu et al. 1998) facilitate structural and mechanistic interpretation of the AAA+multiple alignment. Furthermore, publication of the NSF–D2 structure also provides independent confirmation of the structural similarity between the RFC and AAA families that was predicted by our sequence analysis (presented at a New York Structural Biology Group meeting at Cold Spring Harbor Laboratory, July, 1998). The predicted common structural core shared by these two proteins is quite striking (Fig.2), despite their lack of significant pairwise sequence similarity (Fig. 1).
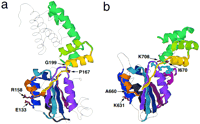
Core structural components corresponding to conserved regions in the AAA+ multiple alignment. (a) Structural components of the DNA polymerase δ′ subunit (pdb: 1A5T). (b) Structural components of NSF–D2 (pdb: 1NSF). Four key conserved positions are indicated. (1) E133/K631 and (2) R158/A660 correspond to conserved (charged residue) positions in boxes VI and VII, respectively (see Fig.4 and text). (3) P167/I670 corresponds to a conserved hydrophobic position in box VII′ that presumably establishes an important contact with the amino-terminal region of the AAA+ module (Yu et al. 1998); in both Pol III d′ and NSF–D2 this hydrophobic residue contacts a Trp residue. (4) G199/K708 corresponds to a conserved basic residue position within the sensor-2 motif (see text). Each core structural element has the same color as the bar over the corresponding aligned region in Fig. 1. Unconserved regions are shown as single gray threads. ATP is colored cyan in b. The β-strand and three-helix bundle corresponding to the last four (orange, yellow, lime, and green) components and the box II (scarlet) component are distinct structural characteristics of the AAA+ class (see Fig. 3and text). Note, however, that the box II component is absent from Pol III δ′. These and the other structural images were created using the RASMOL program by Roger Sayle (GlaxoWellcome).
The AAA+ conserved regions map to five parallel strands that make up a β-sheet and several surrounding helices in a first domain, to a small three-helix bundle that makes up a second domain, and to the first helix of a third domain (Fig. 2). With few exceptions, highly conserved positions correspond to residues that, in the catalytic members of this class, interact with ATP. (Note, however, that the nucleotide binding site in Pol III δ′ is nonfunctional.) Moderately conserved positions generally correspond to interactions within the structural core.
The relationship of the conserved motifs to the Pol III δ′ and NSF–D2 structures can be seen by comparing Figures 1 and 2. The first eight motifs map to the first domain and the last three motifs to a second domain. It seems preferable, however, to combine the last four motifs into one structural group, even though only the last three correspond to domain 2. The reason for this is that the Walker A to sensor-1 motifs correspond to an α,β-fold structural arrangement that is generally similar to the RecA ATP-binding domain (Story and Steitz 1992) found in other ATPases. In contrast, these last four motifs beyond the sensor-1 motif correspond to a distinct structural feature of the AAA+ class.
The first (RFC box II) motif is another distinguishing feature of this class, though it is not always conserved. It is absent, for instance, from the Pol III δ′ subunit but appears to be present in NSF–D2, though its level of sequence similarity to other AAA+proteins in this region is weak. NSF–D2 residues corresponding to the carboxy-terminal region of this motif are in the vicinity of the adenine group of ATP, suggesting a role in adenine recognition (Guenther et al. 1997; Lenzen et al. 1998).
The next six motifs correspond to a P-loop α,β-fold domain. These include the Walker A motif (RFC box III) involved in the phosphate binding of ATP (Walker et al. 1982; Saraste et al. 1990), the Walker B motif (RFC box V) involved in metal binding and ATP catalysis, and the the sensor-1 motif (motif C; Koonin 1993a), which includes a conserved Asn or Thr position that could hydrogen bond with the terminal phosphate of ATP and thereby detect nucleotide binding or hydrolysis (Guenther et al. 1997). The sensor-1 motif may structurally and functionally correspond to motif IV seen in the vast class of ABC-type ATPases (Gorbalenya and Koonin 1990) and to analogous motifs seen in other ATPases (Story and Steitz 1992; Subramanya et al. 1996). The box VI motif, which is located between the Walker B and sensor-1 motif, is associated with interactions between adjacent subunits in NSF–D2 (see below).
The four carboxy-terminal motifs correspond to box VII and the sensor-2 motif as well as to two more subtle motifs (boxes VII′ and VII′′) that to our knowledge have not been reported previously. In Pol III δ′ these four motifs form a connecting link between domains 1 and 3. The box VII motif often contains a conserved Arg residue that in Pol III δ′ occurs at the amino-terminal end of the β-strand directly joined to domain 2 (Fig. 2a). This strand appears to form a lever capable of repositioning domains 2 and 3 relative to domain 1, perhaps upon interaction of the conserved Arg with the nucleotide phosphate group of an adjacent hexameric subunit (see below).
The three-helix bundle of domain 2 maps to the next three motifs. The last of these is the sensor-2 (or box VIII) motif, which is characterized by a highly conserved Arg residue. The corresponding residue at this position in NSF–D2, which is a Lys rather than an Arg, binds to the phosphate group of ATP (Fig. 2b). Based on studies of other ATPases, binding of a basic residue in this way may induce a conformational change that shields the catalytic site from water (seeGuenther et al. 1997 for references). This region may also be involved directly in protein-substrate remodeling, considering that mutations corresponding to the sensor-2 motif of the yeast RFC1 clamp-loader subunit can be rescued by mutations in the PCNA clamp (McAlear et al. 1994). Similarly, in the Pol III γ-subunit (DP3X_ECOLI in Fig. 1) an ATP-induced conformational change, which facilitates interaction between the clamp-loader complex, the DNA clamp and DNA, involves two Arg residues within the box VII′′ and sensor II motifs (Hingorani and O’Donnell 1998).
The hexameric structure of NSF–D2 provides further insight into the relationship between the P-loop α,β-fold domains and the AAA+-specific structural components. The hexameric P-loop domains appear to serve as a platform upon which the AAA+-specific components are mounted (Fig. 3). The latter consists of the box II regions, which appear to serve as lids over the ATP-binding pockets (although their locations in NSF are somewhat uncertain because of sequence divergence); and the box VII to sensor-2 regions, which form knob-like projections. These projections are positioned strategically relative to the bound ATPs and are linked to one another within the hexameric structure (Fig. 3), presumably thereby providing a mechanism to couple ATP binding or hydrolysis to substrate remodeling.
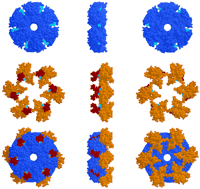
The relationship between P-loop and AAA+-specific structural components within the NSF–D2 hexamer (pdb: 1D2N). Images in the first to third rows correspond to (1) α,β-fold P-loop components (bluish gray), (2) AAA+-specific components (amino-terminal box II regions in red and carboxy-terminal box VII to sensor-2 regions in orange), and (3) all components. (The location of the box II element of NSF–D2 lacks strong statistical support and should be considered tentative.) Images in the first to third columns correspond to view (1) from the surface of NSF–D2 that faces the D1 domain, view (2) from the side, and view (3) from the carboxy-terminal surface. ATP is colored cyan. (See text for details.)
Close examination of the multiple sequence alignment in light of the hexameric structure is also revealing. For example, a Lys residue at position 631 of NSF, which aligns with the highly conserved rightmost position of box VI, contacts the phosphate group of an ATP bound to an adjacent subunit (Fig. 4a). In many AAA+ modules this position corresponds to an acidic residue, such as Glu in Pol III δ′ or Asp in NSF–D1. An acidic residue at this location in the structure may influence positioning of an adjacent subunit and its bound ATP by placing a negative charge near ATP phosphate groups and the coordinated Mg2+ ion. Likewise, an Ala residue at position 660 of NSF–D2 aligns with a highly conserved position within box VII that most often corresponds to a basic residue (colored magenta in Fig. 1). A basic residue at this location may also link binding or hydrolysis of ATP to conformation changes by interacting with a phosphate group. Both of these hypothetical interactions are modeled for Pol III δ′ in Figure 4b. Such interactions may provide a link between adjacent subunits and also couple ATP binding or hydrolysis to conformational changes that could be propagated to carboxy-terminal domains by the box VII–VII′ β-strand.
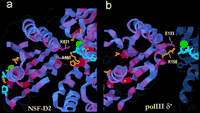
Residues at conserved positions within box VI and VII that make known or potential interactions with an ATP bound to an adjacent subunit. (a) Empirically determined contact within NSF–D2 between Lys-631 and an ATP phosphate group. (b) Hypothetical interaction between Glu-133 and Arg-158 of Pol III δ′ and an adjacent ATP magnesium ion and phosphate group. (See text for residue positions in the alignment.) Color scheme: Residue positions are shaded red proportional to the degree of sequence conservation in the alignment, residue side chains for highly conserved positions are orange, ATP is cyan, and magnesium is green.
Distribution of the AAA+ Class Members in Complete Genomes
Whole-genome analysis indicates that the AAA+ class is ancient and has undergone considerable functional divergence prior to the emergence of the major divisions of life, namely bacteria, archaea, and eukaryotes (Table 1). Furthermore, two distinct groups within this class also span the three major divisions. Other groups, however, have a more patchy distribution and can be classified into two types: those shared by two divisions of life and those specific to one division. As horizontal gene transfer blurs these boundaries, assignment to a division of life was based on the presence of a given group of ATPases across a wide phylogenetic range of species from a given division.
Whole-Genome Analysis of AAA+Proteins
Universally Conserved Groups
There are two groups within the AAA+ class that appear to be represented in all complete genomes sequenced to date: the classic AAA family proteins and the RFC-related clamp-loader subunits (Table 1). Furthermore, some subfamilies within the AAA family are represented in all three major divisions of life. For example, the Cdc48 subfamily, which has two ATPase domains, is conserved in some bacteria, such asMycobacterium tuberculosis, and in all eukaryotes and archaea. In contrast, the FtsH subfamily, which has an additional metalloprotease domain, is highly conserved among bacteria and eukaryotes but is not found in archaea, suggesting that eukaryotes may have acquired this protein from their endosymbionts. The eukaryotes also show an expansion of a universal subfamily consisting of 26S proteasomal regulatory subunits, which have a single ATPase domain. The consistent linkage of this family with protein degradation suggests that even in the common ancestor of all organisms, they may have served as chaperones assisting in protein unfolding and degradation. Some members of the RFC family in eukaryotes have acquired other functions, such as the Schizosaccharomyces pombe Rad17 protein that functions in DNA damage checkpoint sensing (Lydall et al. 1996). Interestingly, all of the examined bacteria possess a clamp-loader subunit that, as for the E. coli Pol III δ′ subunit, has a disrupted P-loop; this suggests that even in the common ancestor of the bacterial lineage the functional diversification of the clamp loader into active and inactive subunits had occurred.
Families Shared by Two Divisions
Some families have been inherited vertically from the last common ancestor of the archaea and the eukaryotes. One of these is the Orc1/Cdc6 family, some inactive members of which have been recruited for other functions—such as Sir3 that regulates chromatin structure in yeast (Hecht et al. 1995). Interestingly, Methanococcus jannaschii lacks Orc1/Cdc6 family members, suggesting that this family may be dispensable in the archaea. MCM proteins constitute another such vertically inherited family. Each of the sequenced archaeal genomes encodes one member of this family except forMethanococcus, which has three; in eukaryotes this family appears to have expanded very early in their evolution to six proteins that have been conserved vertically ever since.
Some families, such as ClpA/B, ClpX/Y, and Lon, appear to have entered eukaryotic genomes by horizontal transfer from bacterial endosymbionts, which subsequently evolved into mitochondria. Similarly, the ClpA/B family in Methanobacterium appears to have been acquired directly from the bacteria through horizontal transfer. The distribution of other families shared by the bacteria and the archaeae, such as the Mg2+ chelatases and the MoxR-related proteins, clearly suggest multiple, probably independent horizontal transfers during evolution (Table 1).
Division-Specific Families
There are several families of AAA+ ATPases that appear to be limited to bacteria. For instance, DnaA, which plays an indispensable role in replication, occurs in at least one copy in all of the bacterial genomes sampled thus far; E. coli andHaemophilus influenzae encode a second, truncated copy of DnaA, which may be inactive in E. coli. RuvB helicase is also widely represented in the bacteria and is missing only in the extreme thermophile Aquifex aeolicus. The nitrogen transcription regulatory protein C (NtrC) family shows a patchy distribution in bacteria and is seen in distantly related branches such asAquifex, E. coli, and Bacillus in 4–10 copies per genome (Table 1). The expansion of this family may correlate with the presence of its functional partner, the σ-factor RpoN. There are some other families that, thus far, are restricted to single bacterial species, such as SpoVJ in Bacillus andMycobacterium.
The most striking family specific to the archaea is one in which a Lon-like serine protease domain is fused to an AAA+ module. This appears to represent a novel archaeal ATP-dependent protease that is likely to be mechanistically similar to the Lon proteases.
Eukaryotes also appear to have evolved new families, including the giant protein dynein that has six AAA+ modules. The origin of such proteins may correlate with the evolution of the eukaryotic cytoskeleton. There is a similarly huge protein with six ATPase domains in yeast (the largest protein encoded in its genome) (Fig. 1) whose, probably important, function is still unknown.
Domains Covalently Linked to AAA+ Modules
AAA+ modules are often linked covalently to other domains that may provide valuable clues about their cellular functions. These domains fall into two categories: protease domains and interaction domains (Fig. 5). The former includes both serine proteases and metalloproteases. Because AAA+ modules often participate in the assembly of protein complexes, these protease domains were most likely acquired to help degrade misfolded components that fail to integrate properly. Apparently a serine protease domain has become associated with an AAA+ module on at least three different occasions in the course of evolution. This seems likely because a serine protease is fused to the amino terminus of some AAA+ modules but to the carboxyl terminus of others, and the corresponding AAA+ modules cluster into three distinct sequence similarity groups.
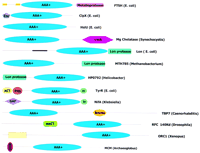
Schematic representations of the domain architectures of AAA+proteins. (Znc) 5 cysteine zinc cluster; (vWA) von Willebrand factor A domain; (ACT) novel ligand binding domain; (H) helix–turn–helix DNA-binding domain; (BRCT) BRCA carboxy-terminal domain; (bromo) bromodomain. Domains references can be found at the SMART Web site (Schultz et al. 1998). The yellow box in ORC1 represents a novel uncharacterized domain seen in chromatinic proteins. Coiled–coil regions were predicted using the Coils program (Lupas 1996); signal peptides and transmembrane regions were predicted using Signal-P (Nielsen et al. 1997) and PHD-Tmpred (Rost et al. 1996), respectively.
Other domains covalently linked to AAA+ modules mediate localization to cellular membranes or interaction with nucleic acids or other proteins. These interactions may provide functional specificity or help target the protein to specific cellular sites. For example, a helix–turn–helix DNA-binding domain targets NtrC-like proteins to specific DNA sequences. Moreover, some of these σ-dependent transcriptional regulators also possess ligand-binding domains, such as PAS (Ponting and Aravind 1997) and GAF (Aravind and Ponting 1997), which may regulate ATPase activity in a ligand-dependent manner. Eukaryotic RFC proteins possess a BRCT domain (Koonin et al. 1996a), which is a common module seen in DNA repair and checkpoint regulator proteins. This domain is likely to participate in homophilic protein–protein interactions, thereby recruiting other proteins containing BRCT domains.
DISCUSSION
The AAA+ class is a collection of chaperone-like modules that appear to function as molecular matchmakers in the assembly, operation, and disassembly of diverse protein machines. Many of the ATPases in this class are known chaperones, and many others serve as molecular matchmakers in the formation or activation of DNA–protein complexes. In the latter case, the hexameric architecture often associated with this class can provide a hole through which DNA may be thread, thereby anchoring the complex to DNA. Moreover, given the strong association of this class with known chaperones, it is important to understand how protein remodeling may play a role in those cellular activities not linked previously to chaperones, but now found to involve members of this class.
AAA+ Chaperones
Many of the known AAA+ chaperones perform similar and sometimes overlapping functions. For example, overproduction of yeast Lon promotes mitochondrial respiratory complex assembly in cells lacking Afg3p and Rca1p (Rep et al. 1996; Suzuki et al. 1997), which are members of the AAA family that normally perform this function. This is consistent with the notion that the Lon and AAA proteases are evolutionarily, structurally, and functionally related. Likewise, overexpression of the ClpYQ protease complex in E. colisuppresses the SOS-mediated inhibition of cell division seen inlon mutants (Khattar 1997). Moreover, Clp protease and ATPase subunits form cylindrical four-ring complexes that resemble the eukaryotic 26S proteasome (Kessel et al. 1995; Goldberg et al. 1997;Rohrwild et al. 1997). Note, however, that the chaperone function of at least one Clp family member, Hsp104, is unrelated to proteolysis (Glover and Lindquist 1998).
AAA+ Modules Associated with Protein–DNA Complexes
There are mechanistic similarities between DNA replication, transcription, and recombination (Kodadek 1998). In particular, for all of these activities, the high specificity and stability needed to establish an initial DNA–protein complex conflicts with the flexible state of processive activity these complexes assume while performing their particular functions (Dutta and Bell 1997; Baker and Bell 1998). For this reason, both the assembly of an initial complex and the subsequent transition to and maintenance of an active protein machine are likely to require subunit remodeling, which appears to be mediated by these chaperone-like AAA+ modules.
AAA+ modules are found in many proteins associated with the initiation of DNA replication. In yeast, these include the Orc1, Orc4, and Orc5 subunits of the origin recognition complex (ORC) (Bell et al. 1993), the Cdc6 protein (Liang et al. 1995), minichromosome maintenance (MCM) proteins (Chong et al. 1996), and RFC family members (Cullmann et al. 1995). These are involved in successive steps in the initiation of replication (for reviews, see Kelman and O’Donnell 1995; Chong et al. 1996; Kearsey et al. 1996; Stillman 1996; Rowles and Blow 1997; Toone et al. 1997). ORC is required for Cdc6 binding to chromatin, ORC and Cdc6 are required for MCM binding, and later the RFC complex is required to load the PCNA sliding clamp onto DNA. The similarity of these ATPase modules to known chaperones suggests that their role is to remodel and load protein subunits, which may also harbor similar ATPase modules that can then load still other subunits and so on, until the entire initiation complex is assembled. Some functionally analogous bacterial proteins, such as DnaA, also contain these modules. Viral proteins with functions analogous to that of MCM proteins and DnaA, such as SV40 large T antigen (Roberts 1989), are distant relatives of the AAA+ class (Koonin 1993a; L. Aravind, E.V. Koonin, and A.F. Neuwald, unpubl.).
AAA+ modules are also associated with transcription factors related to the bacterial NtrC protein. NtrC activates transcription from a distant enhancer DNA sequence by remodeling the closed complex between promoter DNA and RNA polymerase to an open complex (Wyman et al. 1997and references therein). Likewise, several eukaryotic members of the AAA+ class, such as the Tip49 protein (Kanemaki et al. 1997), function as transcription factors. Interestingly, DnaA, an AAA+protein normally associated with the initiation of DNA replication, also functions as a transcription factor (for review, see Messer and Weigel 1997), again suggesting that similar remodeling mechanisms may be involved in the initiation of both DNA replication and transcription.
RuvB, a motor protein that promotes DNA branch migration at Holliday junctions during genetic recombination (Rice et al. 1997; West 1997), also contains one of these ATPase modules. RuvB works in concert with RuvA, with which it forms a complex consisting of RuvA sandwiched between two RuvB hexameric rings (Yu et al. 1997). DNA is thread through a hole in these rings. RuvB’s similarity to chaperones suggests that it may induce conformational changes in RuvA, perhaps leading to a rachet-like movement of RuvA “acidic pins” at the junction point to facilitate DNA unpairing and strand migration (Rafferty et al. 1996). At the same time a conformational change in the RuvB hexamer itself, which possesses helicase activity, could directly assist in DNA rotation and translocation through this complex. Notably, helicase activity has also been reported for other AAA+ proteins, including MCMs (Ishimi 1997) and SUG1 (Fraser et al. 1997). And, as for known helicases (Wessel et al. 1992; Hacker and Johnson 1997; Martin et al. 1998), many ATPases in this class are components of hexameric complexes; Lon (Kutejova et al. 1993) and MCMs (Ishimi 1997) being two additional examples.
The B subunit of the restriction endonuclease McrBC contains one of these AAA+ modules, which, unlike other such modules, is a GTPase. This endonuclease recognizes and cleaves a relatively extensive region of DNA, up to 80 bases or more and, therefore, may form an initiation complex prior to its activation. If so, then—as was suggested recently (Gast et al. 1997; Pieper et al. 1997)—this GTPase module may be involved in the transition from initial DNA binding to a catalytically active endonuclease.
AAA+ Chaperones Associated with DNA–Protein Complexes
If these AAA+ modules do perform remodeling functions associated with DNA–protein complexes, it is not surprising that these ATPases share sequence similarity to known chaperones or, conversely, that known chaperones are sometimes involved in the assembly of DNA–protein complexes. ClpA, for example, can induce the in vitro activation of the bacteriophage P1 replication initiator protein RepA (Wickner et al. 1994). Hexameric ClpA accomplishes this by remodeling RepA dimers into monomers (Pak and Wickner 1997), thereby stimulating RepA’s DNA-binding activity (Wickner et al. 1991). Hence, in a rather simple way, ClpA seems functionally analogous to homologous ATPases that serve as DNA replication initiation factors. Similarly, ClpX alters the conformation of MuA to promote the transition from a stable MuA–DNA complex to DNA synthesis during bacteriophage Mu DNA replication by transposition (see Jones et al. 1998 and references therein). Human Lon also binds specifically to single-stranded DNA in a region of the mitochondrial genome involved in regulation of DNA replication and transcription (Fu and Markovitz 1998), suggesting that it may target and remodel specific DNA-binding proteins either for selective degradation or for assembly. Furthermore, the bacterial Lon protein has nonspecific DNA-binding activity (Charette et al. 1981; Zehnbauer et al. 1981), and its protease activity is stimulated by DNA (Charette et al. 1984), suggesting functional similarity to other DNA-binding members of the AAA+ class.
Transcription-related functions have been associated with regulatory components of the 26S proteasome (for references, see Baumeister et al. 1998). For example, human SUG1 interacts directly with a subunit of the transcription initiation and DNA repair factor TFIIH (Weeda et al. 1997). This interaction appears unrelated to proteolysis of TFIIH, which has lead to the suggestion that SUG1 may remodel RNA Pol II to free this factor from the transcriptional machinery for use by the repair machinery (Weeda et al. 1997). Similarly, the yeast SUG1 protein has been associated with the RNA Pol II holoenzyme (Kim et al. 1994), suggesting a transcriptional role. Because SUG1 is also a proteasome component, this implies dual degradative and assembly roles similar to that noted for the AAA proteins Afg3p and Rca1p (Weeda et al. 1997). Conversely, as suggested by Dubiel et al. (1992), AAA family members not associated currently with the proteasome may, in fact, also function as proteasome components. This has been borne out by several studies, including the recent finding that valosin-containing protein (VCP), a mammalian protein associated with membrane fusion, is involved in ubiquitin-proteasome-mediated degradation of IκBα, an inhibitor of the transcription factor NF-κB (Dai et al. 1998). Consistent with a role for some AAA+ ATPases in transcription regulation at the chromatin level, we observed that in the yeast protein TBP-7 and in its ortholog from C. elegans, the AAA+module is fused to a bromodomain (Fig. 5), which suggests a possible role for this ATPase in chromatin remodeling.
AAA+ Modules Associated with Other Functions
The functions associated with other AAA+ families that appear unrelated to DNA binding or proteolysis may also involve chaperone remodeling and assembly or activation of protein complexes. For example, one of these ATPase module occurs in Rubisco activase, which couples ATP hydrolysis to the release of inhibitory sugar phosphates bound to Rubisco active sites (Salvucci and Ogren 1996). Consistent with its sequence similarity to known chaperones, it has been suggested, based on experimental evidence, that Rubisco activase functions as a chaperone rather than a conventional enzyme (Sanchez de Jimenez et al. 1995). AAA+ modules are also found in the Mg2+ chelatase complex, which requires an ATP-dependent activation step prior to insertion of Mg2+ into the precursor of bacteriochlorophyll (for review, see Walker and Willows 1997). This activation step may be analogous to the priming of SNARE proteins by NSF, whereby the AAA+ module remodels protein subunits to prime them for the Mg2+ insertion step. Cytoplasmic dynein, which acts as a motor for the transport of membranous organelles along microtubules, contains six of these ATPase modules that appear to be associated with its motor activity (for reviews, see Ogawa and Mohri 1996; Vallee and Sheetz 1996; Hirokawa 1998). Given that (as shown here) dynein is a homolog of RuvB, both of these motor proteins may share similar mechanisms, perhaps involving iterative rounds of chaperone-like remodeling.
Additional cellular activities associated with AAA+ modules are likely to emerge upon characterization of the many hypothetical and poorly understood proteins detected in this class. Of these, some of the human proteins may be associated with genetic diseases considering that class members are often involved in essential functions. One such protein, noted previously to share sequence similarity to Clp ATPases (Ozelius et al. 1997), is TorsinA, which is mutated in early-onset torsion dystonia—a movement disorder characterized by twisting muscle contractures. Intriguingly, TorsinA mutations appear to cause a defect in release of dopamine, rather than a defect in dopamine synthesis (Ozelius et al. 1997 and references therein). Thus, as for dynein, TorsinA may function as a motor protein in the transport of dopamine-containing membranous vesicles. Alternatively, TorsinA could perform a role similar to that of NSF in vesicle-membrane fusion.
Just as some manmade devices, such as the electric fan, are components in a disproportionate number of machines, the AAA+ module plays a role in a disproportionate number of cellular activities. These modules consist of a P-loop ATPase motor domain upon which an AAA+-specific component is mounted. What appears to make this module so useful for so many cellular activities is its ability to interact both with nucleic acids and proteins and to either assemble or reshape molecular complexes or to dismantle them through protein degradation.
METHODS
Detection and Alignment of AAA+ Proteins
PROBE (Neuwald et al. 1997) was used to obtain a multiple sequence alignment of the AAA+ class. PROBE relies on iterative database search and multiple alignment steps to detect and align class members until convergence. Additional relationships were detected using PSI-BLAST (Altschul et al. 1997). During multiple alignment, PROBE stochastically searches for an optimal alignment using a genetic algorithm, and hidden Markov model and Gibbs sampling methods. Conserved patterns are located using a statistical criterion (Neuwald et al. 1997) that specifies how many ungapped conserved regions (or blocks) and which positions within each block to include in the alignment model. As a result, an optimum model represents only those aspects of the aligned sequences showing some evidence of shared functional constraints.
The detection and correct alignment of AAA+ proteins required the following modifications of the PROBE program. First, when computing the statistical significance of matches to multiple alignment profiles of these sequences, the highly conserved positions corresponding to the Walker A and B motifs [“(ILVF).G..G.GK(ST)” and “DE..”, respectively] were ignored. This was done by setting the scores at these positions to zero when determining statistical significance. This avoided detection and inclusion of otherwise unrelated ATP-binding proteins in the final alignment. To increase sensitivity further, database searches relied on a new statistical procedure that takes into consideration the gaps between multiply aligned conserved regions (described below). PROBE was also modified to detect and align repetitive domains in individual sequences.
Several new optimization procedures were also added to the PROBE multiple alignment method. Near-optimum sampling (Neuwald et al. 1995) and simulated annealing procedures were incorporated into propagation, the hidden Markov model version of Gibbs sampling used by PROBE (Liu and Lawrence 1995; Neuwald et al. 1997). Both of these procedures can improve the alignment after convergence by attracting the Gibbs sampler to a local optimum. Several new Gibbs sampling procedures were also added to facilitate escape from suboptimal kinetic traps. All of these optimization procedures (A.F. Neuwald, unpubl.), have no effect on the validity or nature of the alignment only on the speed with which an optimum is found.
To detect and eliminate false positives, the following “jackknife” statistical procedure was applied. First, homologous “domains” aligned by PROBE (with flanking regions removed) were clustered into groups sharing significant transitive pairwise sequence similarity (E ≤ 0.01 after an adjustment for the size of the protein database). Then, for each group except for the main group, it was determined whether a PROBE alignment model of the remaining sequences detects at least one sequence in that group in a search of the entire nonredundant database (P ≤ 0.01). If not, that group was assumed to contain false positives and was discarded from the alignment set. Several borderline relationships were validated through PSI-BLAST searches.
Alignment with Gap Functions and Short Insertions and Deletions
Conserved regions in a multiple alignment are separated typically by unconserved regions that are best left unaligned. Yet, even though the regions themselves may be unconserved, their lengths may be conserved to varying degrees for a particular protein family. If so, then the sensitivity of a profile search may be improved by modeling the lengths of these unconserved regions, which we call gaps. To do this, empirical likelihood estimates of gap propensities were determined and incorporated into alignment profiles and corresponding statistical procedures were devised.
Log-odds gap scores were estimated directly from PROBE multiple sequence alignments as follows. First, a score was obtained for each possible gap length from its empirical frequency, or more exactly, from the likelihood-ratio of its empirical frequency over a uniform gap frequency. Then a standard smoothing function (Savitzky and Golay 1964) was applied to these ratios and integer scores were obtained by rounding the (natural) logarithms of these values. This smoothing function adjusts for estimation errors caused by small sample size. Optimal sequence-to-model alignments are obtained during a database search using a standard dynamic programming procedure based on the gap and the residue substitution scores.
The statistical significance of these gapped alignment profile scores were assessed using the following procedure (the mathematical and algorithmic details of which will be presented elsewhere) (J.L. Spouge, unpubl.). This procedure is a generalization of an efficient recursive procedure described by Staden (1989). The Staden procedure adds up the probabilities associated with specific integer alignment scores, given particular amino acid background frequencies and an ungapped position-specific scoring matrix. This procedure has been incorporated into the PROBE database search step (Neuwald et al. 1997). The new procedure differs in that it also takes into account the specific log-odds gap scores. Note that the gaps occur between (ungapped) aligned segments and that overlapping segments are prohibited. To see how the original Staden procedure can be extended in this way, gaps of different lengths should be viewed as additional symbols in the residue alphabet. The length of the aligned sequence restricts the possible gap lengths, however, and the modified calculation needs to account for this. The code performing the modified calculation was verified numerically; analytically, it is known to provide conservative Bonferroni (Galambos 1975, 1977) P-value estimates.
The multiple alignment algorithm was also modified to accommodate short insertions and deletions within conserved regions. This modified version uses a dynamic programming procedure with affine gap penalties to detect insertions and deletions in each sequence relative to the alignment model. To prevent unwarranted gapping, conservative gap penalties were used and—rather than probabilistically sampling from among all possible gapped alignments—only the best alignment was selected. (A full Gibbs sampling version of this procedure is currently being developed; A.F. Neuwald, unpubl.) Next, sequences were realigned using a modified version of the PROBE alignment algorithm that accommodates these insertions and deletions. This entire procedure was then iterated by again applying the dynamic programming step to detect short insertions and deletions relative to this new alignment model, followed by another realignment, and so on until convergence (that is, until the current alignment was nearly identical to the alignment obtained in the previous iteration). This gapping procedure was applied only after convergence on an ungapped multiple alignment.
Whole-Genome Analysis
Sequence clusters used for this analysis were based on a single-linkage clustering procedure (Koonin et al. 1996b) with serial bit cutoff scores from 40 to 70 reduced in units of 5. This procedure ensures that proteins are grouped into distinct clusters that are not altered easily by slight changes in cutoff scores. Robustness of the clusters was verified by showing that, in PSI-BLAST searches of the NR database, the highest scoring hits for members of each cluster were other members of that cluster. Only representatives from complete genomes were used for clustering.
Acknowledgments
We thank Bruce Stillman for suggesting an analysis of RFC-related proteins and for insightful comments and James Chong for critical reading of the manuscript and helpful suggestions. A.F.N. was supported in part by grant 5P30 CA45508-11 from the National Cancer Institute and grant 1R01 LM06747-01 from the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Corresponding author.
-
E-MAIL neuwald{at}cshl.org; FAX (516) 367-8461.
-
- Received November 6, 1998.
- Accepted December 8, 1998.
- Cold Spring Harbor Laboratory Press








