
Large-Scale Taxonomic Profiling of Eukaryotic Model Organisms: A Comparison of Orthologous Proteins Encoded by the Human, Fly, Nematode, and Yeast Genomes
Abstract
Comparisons of DNA and protein sequences between humans and model organisms, including the yeast Saccharomyces cerevisiae, the nematode Caenorhabditis elegans, and the fruit flyDrosophila melanogaster, are a significant source of information about the function of human genes and proteins in both normal and disease states. Important questions regarding cross-species sequence comparison remain unanswered, including (1) the fraction of the metabolic, signaling, and regulatory pathways that is shared by humans and the various model organisms; and (2) the validity of functional inferences based on sequence homology. We addressed these questions by analyzing the available fractions of human, fly, nematode, and yeast genomes for orthologous protein-coding genes, applying strict criteria to distinguish between candidate orthologous and paralogous proteins. Forty-two quartets of proteins could be identified as candidate orthologs. Twenty-four Drosophila protein sequences were more similar to their human orthologs than the corresponding nematode proteins. Analysis of sequence substitutions and evolutionary distances in this data set revealed that most C. elegans genes are evolving more rapidly than Drosophila genes, suggesting that unequal evolutionary rates may contribute to the differences in similarity to human protein sequences. The available fraction ofDrosophila proteins appears to lack representatives of many protein families and domains, reflecting the relative paucity of genomic data from this species.
Similarities between novel protein sequences and their better-characterized counterparts in sequence databases are an increasingly important source of hypotheses concerning protein functions. Particular attention has been paid to identifying homologs of medically relevant human proteins in genetically tractable model organisms, such as mice, the fruit flyDrosophila melanogaster, the nematode Caenorhabditis elegans, the yeast Saccharomyces cerevesiae, and bacteria (Banfi et al. 1996; Bassett et al. 1997; Mushegian et al. 1997). Whole-genome comparisons of microbial proteins (Koonin et al. 1997) have emphasized the importance of distinguishing orthologs, that is, proteins in two species that have evolved by vertical descent from a common ancestor and are presumed to have the same function (Fitch 1970), from paralogs, namely proteins derived from lineage-specific duplication and domain shuffling that hence may have more divergent functions. Failure to resolve orthologs and paralogs can lead to misinterpretation of cellular biochemistry (Tatusov et al. 1996;Henikoff et al. 1997) and inaccuracies in molecular evolutionary reconstructions (Doolittle et al. 1996; Feng et al. 1997). This distinction has been addressed in a protein domain analysis of positionally cloned human genes that are mutated in specific diseases (“disease genes”) and their counterparts in the yeast and nematode genomes (Mushegian et al. 1997). Although almost equal fractions of the human disease genes had regions of significant similarity to nematode and to yeast proteins, the latter study identified a true ortholog in the complete yeast proteome for only 20% of human proteins. In contrast, 30% of human disease genes had candidate orthologs in the ∼50% completed nematode proteome then available.
Drosophila and C. elegans have emerged as attractive model animal systems for studying human gene pathways, because of their genetic tractability, phenotypically well-characterized genes, and progress in whole-genome sequencing (Rubin 1996; Ahringer 1997). Traditional morphology-based phylogenies have placed C. elegans and other nematodes in a basal metazoan clade composed of pseudocoelomate animals, whereas Drosophila and other arthropods have been placed in the more recently derived protostome clade of eucoelomate animals with a shorter phylogenetic distance to vertebrates and other deuterostomes (for review, see Brusca and Brusca 1989). However, the notion that arthropods belong to a later evolutionary branch than nematodes (e.g., Sidow and Thomas 1994) has been challenged by recent studies based on analysis of morphology (Nielsen 1995), of ribosomal RNA sequences (Aguinaldo et al. 1997), and of selected protein sequences (McHugh 1997). Notably, the estimated sizes of the Drosophila and C. elegans genomes and proteomes are quite similar, on the order of 100 MB of genomic DNA and 15,000 genes (Miklos and Rubin 1996; Waterston 1997). Despite this information, the representation of proteins and conserved protein domains in these two proteomes has not been approached systematically.
The present study was designed to identify a substantial set of orthologous protein-coding genes in the eukaryotic model organisms by using strict criteria to define orthologous candidates. We then analyzed this set of proteins to assess the relative similarity ofDrosophila and C. elegans proteins to their human orthologs. As a complementary approach toward the evaluation of the model organisms, we sought to estimate the fractions of conserved domains and to compare the composition of multidomain proteins in the available protein sets of Drosophila and C. elegans.
RESULTS AND DISCUSSION
Forty-Two Quartets of Candidate Orthologs
To identify all potential orthologous genes in humans, nematode, fly, and yeast, we first searched the complete S. cerevesiaeproteome (6141 protein sequences) using as queries identifiedDrosophila proteins (2142 available sequences as of March 1, 1997, then excluding peptides shorter than 110 amino acids). The modified BLATAX program was used to tabulate the highest scoring matches for each Drosophila protein. The resultant 848 fly proteins were then used to search the nonredundant protein sequence (NR) database and extract the best matches from humans. The human sequences retrieved in this way were examined to remove incomplete proteins and sequences that occurred more than once as a result of being the best match for two or more Drosophila proteins. The remaining set of 480 human proteins was used to search the NR database. The best matches to these human proteins from Drosophila, C. elegans, and S. cerevesiae were then extracted by the modified BLATAX program using the similarity score measure. Next, we removed low-scoring sets and spurious hits representing matches in low-sequence complexity and coiled–coil segments. The resultant set of proteins was filtered to derive candidate orthologs by excluding (1) sequences for which the yeast sequence was closer to a human homolog than either a fly or nematode sequence (the third criterion of orthology, see Methods); and (2) proteins that shared high similarity in one domain but differed in overall domain architecture from the human protein (the second criterion of orthology). We then excluded members of expanded protein families as listed in Methods. The remaining protein quartets were subjected to a final round of filtering based on reciprocal BLASTP searches (the first criterion of orthology).
The resulting set of proteins contained 42 quartets of human,Drosophila, C. elegans, and S. cerevesiae candidate orthologs (Table 1). The 42 proteins comprising the data set of orthologs varied extensively in length, for example, the human proteins contained 116–2225 amino acids, with a median of 349 residues. This is a significantly broader spectrum of sizes than the set of 64 enzymes (151–935 residues) used in a recent large-scale phylogenetic comparison (Doolittle et al. 1996). Moreover, the present set of candidate orthologs samples many of the functional categories essential in the eukaryotic cell, including genome replication and expression, organelle structural components, and signal transduction (Table 1).
Forty-Two Quartets of Orthologous Proteins
Different Proteins Generate Different Phylogenetic Tree Topologies
Most of C. elegans proteins in the databases, and 33 of 42 nematode proteins in current data set, are predicted from the genomic sequence, whereas all 42 of the Drosophila orthologs were derived from full-length cDNA sequences. Therefore, additional measures were taken to verify the orthologous candidates. First, the human protein sequences were used as queries to search the database of unfinished nematode DNA for possible missed exons. Second, the EST databases were searched for the higher scoring sequences in the nematode and fly. Only one nematode sequence (synaptobrevin) was found among the ESTs that was a better orthologous candidate than the sequence in the NR database, and was included in the analysis.
The possible relationships of the human, nematode, and fly sequences can be described by three different tree topologies (Fig. 1): (1) tree A, in which the fly sequence is a sister taxon to the human sequence with the nematode sequence basal to the fly–human clade; (2) tree B, wherein the fly and nematode sequences are sister taxa; and (3) tree C, in which the nematode and human sequences are sister taxa with the fly sequence basal to the nematode–human clade.
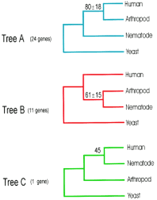
The three possible topologies for a tree describing the evolutionary relationships between nematodes, arthropods, and humans. Tree A (blue) reflects the conventional interpretation of metazoan phylogeny with nematodes as a “protocoelomate” group basal to arthropods and humans. Tree A was supported by neighbor-joining analysis of 24 protein quartets as described in the text. Tree B (red) represents the “Ecdysozoa” phylogeny derived from 18S rRNA gene sequences of a variety of nematodes and arthropods (Aguinaldo et al. 1997), and is supported by 11 protein quartets. Tree C (green) is not expected from any metazoan phylogenetic hypothesis and is supported by a single protein quartet. Average bootstrap values and their standard deviations are shown for each tree.
Thirty-six protein quartets whose metazoan members contained amino acid identities of ⩾35% to the yeast sequence were subjected to individual phylogenetic analysis as described in Methods. Neighbor-joining analyses of the 36 orthologous protein sequence alignments (Fig. 2), revealed that 24 quartets generate tree A (with average bootstrap values of 80% ± 18 s.d.), 11 amino acid alignments support tree B (average bootstrap values of 61% ± 15), and 1 alignment produced tree C with a bootstrap value of 45. Results were essentially identical when gamma-corrected distances were used. These data are consistent with the distribution of the pairwise similarity scores observed in BLAST searches. Eight of the nine C. elegans protein sequences derived from full-length cDNA were less similar to their human orthologs than the corresponding Drosophila protein sequences (i.e., supported tree A), suggesting that the prevalence of Tree A was not an artifact of computational prediction of nematode proteins.
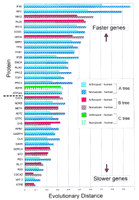
Relative evolutionary rates of the 36 protein quartets subjected to phylogenetic analysis. Protein quartets supporting tree A are shown in blue, those supporting tree B are shown in red, and the quartet supporting tree C is shown in green. The protein name abbreviation is shown along the y-axis, and the proteins are plotted in order of the mean evolutionary distance of nematode to human and arthropod to human where nematode is C. elegans, and arthropod is D. melanogaster. Proteins with the highest number of pairwise substitutions (fast evolving) are at the top and those with the lowest number of pairwise substitutions (slow evolving) are at the bottom. The evolutionary distances along the x-axis were determined from amino acid alignments using a Poisson correction as described in Methods. The broken line (middle left) represents the midway point where 18 proteins are above the line and 18 are below the line. The key to the bars is shown.
Similarity of Fly and Nematode Proteins to Human Orthologs May Be Influenced by Unequal Evolutionary Rate Effects
Phylogenetic hypotheses based on molecular sequence data are affected by two important factors governing evolutionary rate. The first factor is gene-to-gene variation, where different genes have different evolutionary rates among a given pair of taxa, usually as a result of functional constraints on the encoded protein. The second factor is evolutionary rate heterogeneity within a gene, where the evolutionary rate can vary among different lineages of a tree. A homogeneously evolving gene evolves at the same rate per unit time among all branches of a tree, whereas a heterogeneously evolving gene may evolve more rapidly in some lineages than others, causing unequal rate effects that are known to produce tree-building artifacts (Hillis et al. 1994; Lyons-Weiler and Hoelzer 1997; Maley and Marshall 1998). For example, the 18S rRNA gene has evolved at a rapid rate of nucleotide substitution in C. elegans compared to other animals (Winnepenninckx et al. 1995; Garey et al. 1996), causing unequal rate effects that artificially place C. elegans as a basal animal, as in tree A of Figure 1. However, 18S rRNA genes from most nonrhabditid nematode taxa appear to have evolved at a slower rate than in C. elegans (Blaxter et al. 1998), and when nematode 18S rRNA sequences with lower substitution rates are analyzed, nematodes emerge as a sister taxon to arthropods (Aguinaldo et al. 1997), as in tree B of Figure 1.
The finding that only one quartet supported tree C was expected because the possibility that arthropods are basal to both nematodes and humans is not supported by any hypothesis of metazoan phylogeny of which we are aware. The finding that 24 quartets support tree A, whereas 11 quartets support tree B has several possible explanations. One is that tree A reflects the correct historical phylogeny, and that the quartets supporting trees B and C represent random noise. An alternative explanation is that the finding of the majority of quartets supporting tree A is caused by unequal evolutinary rate effects, as in the 18S rRNA gene of C. elegans (Aguinaldo et al. 1997). To assess gene-to-gene variation and evolutionary rate heterogeneity among these 36 orthologous proteins of humans, nematodes, and arthropods, the pairwise evolutionary distances from human to nematode were compared with those of human to arthropod for each quartet. These evolutionary distances are shown as a bar graph in Figure 2, with the quartets ordered from the fastest to slowest evolving proteins. There are fewer pairwise substitutions between arthropod and human than between nematode and human except in five quartets (EF2, SYB, METK, ATPase, PYR1). Figure 2 shows that 8 of 11 quartets that support tree B fall among the slower half of the quartets, whereas only three of the quartets supporting tree B fall among the faster half of the quartets. Five of the eight slowest evolving quartets (ATPB, CDC42, RL17, EF2, SERCA) support tree B. These five proteins have fewer overall substitutions, thus unequal evolutionary rates are less likely to develop and tree B is favored. The faster evolving quartets that support tree B (GPDA, RL22, MA12) likely represent genes that have a high number of substitutions, but where the number of substitutions are homogeneous between the taxa within the quartet.
To visualize the degree of evolutionary rate heterogeneity among all four taxa, we plotted the four-way relative rates for the 36 proteins in Figure 3. The y-axis in Figure 3 displays the ratio of the evolutionary rates of nematode and arthropod relative to yeast, which should equal one if a protein evolved homogenously in the two lineages, as yeast is undoubtedly an outgroup to nematode and arthropod. However, most of the proteins have a y-value greater than one, indicating that they have evolved more quickly in the lineage from yeast to nematode than from yeast to arthropod. Similarly, the majority of the proteins have an x value >1, indicating that most have evolved more quickly in the lineage from human to nematode than from human to arthropod (assuming that nematodes and arthropods are sister taxa). The distribution of data points along thex-axis is highly skewed, with most of the proteins between 0.9 and 1.3, and the remainder with higher values. Of note, proteins supporting tree B are clustered more closely to the point where thex- and y-axes both equal one (representing proteins that evolved homogeneously in all four taxa), but proteins supporting tree A are all to the right with most far to the right (representing proteins that evolved more heterogeneously). Thus, these orthologous proteins can be divided into two populations: homogeneously evolving proteins that support tree B, and heterogeneously evolving proteins that support tree A. This four-way relative rate plot suggests that the preponderance of proteins supporting tree A is largely attributable to unequal evolutionary rates. Given that the orthologous proteins selected for this study were extracted from total available genome data, it is reasonable to infer that approximately two-thirds of protein-coding genes in C. elegans have evolved more rapidly than in Drosophila.
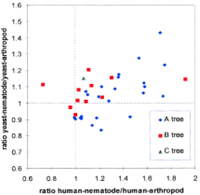
Four-way relative rate plot of evolutionary distances for 36 proteins. The ratio of evolutionary distances from (human–nematode)/(human–arthropod) for each protein is plotted on thex-axis, where a ratio of 1 would be expected if the proteins were evolving homogeneously in those branches assuming that arthropods and nematodes are sister taxa. The ratios of evolutionary distance from (yeast–nematode)/(yeast–arthropod) are plotted on they-axis, which should equal 1 if a protein evolved homogenously in the nematode and arthropod lineages. The position where thex-axis and y-axis both equal one represents the region where genes would fall if they evolved homogeneously in all four taxa, if Tree B is correct. Proteins to the right of the vertical line at x = 1 should favor tree A, proteins to the left should favor tree C, whereas proteins falling near the diagonal line should favor tree B. The distribution of the 36 orthologous proteins is skewed, with those that yield tree B (red squares) scattered uniformly around the diagonal line (with one exception, CDC42 supports tree B but falls to the extreme right of the graph), whereas all of the proteins that yield tree A (blue diamonds) are scattered to the right of the diagonal. The quartet favoring tree C is shown in green (triangle).
Highly Conserved Protein Domains and Diversity of Multidomain Proteins in Drosophila and C. elegans
C. elegans and Drosophila, as the most highly developed genetically tractable model animals, are attractive systems for studying human disease pathways (Miklos and Rubin 1996; Ahringer 1997). In practice, the ability to extract functional inferences depends more on the actual content and complexity of the protein repertoire in different model organisms than on their deduced taxonomic positions per se. To address the question of protein domain diversity in these two proteomes, we first masked putative nonglobular segments and coiled coils in the Drosophila and C. elegansprotein data sets. These two classes of sequences often serve as hinges between globular domains and tend to produce spurious hits with database searches (Altschul et al. 1994). After deleting redundant domains in each database, we constructed Drosophila andC. elegans libraries of unmasked protein domains >50 amino acids and compared these libraries to each other, to the complete yeast proteome, and to the publicly available human ESTs. Matches with a similarity score of >90 were counted, a cutoff that virtually ensures evolutionary relation and functional relevance under the applied conditions (Koonin et al. 1997; A.R. Mushegian, unpubl.).
The results of this analysis are summarized in Table2. The most conspicuous result is that the available portion of the Drosophila proteome is strongly enriched in conserved domains as compared to that of C. elegans. It seems unlikely that Drosophila has retained a larger fraction of proteins descended from an ancestral unicellular eukaryote, while also becoming enriched in protein domains shared with humans. Rather, we suspect that this difference is largely attributable to overrepresentation of certain classes of Drosophila proteins in current databases, given that only a small fraction of the fly genome has been sequenced. A substantial increase inDrosophila genomic DNA sequence will clearly be required before the question of domain repertoire in this organism can be addressed in a more definitive way.
Protein Domain Conservation in Model Organisms
A hallmark of eukaryotic genome evolution is the increased number of multidomain proteins thought to have originated largely by domain shuffling (Doolittle 1995). Because a comprehensive comparison of protein sets of C. elegans and D. melanogaster is limited by the extent of whole-genome sequencing, we wished to analyze a representative set of multidomain proteins in both available proteomes. Toward this end, we extended an earlier analysis of 77 proteins encoded by human positionally cloned genes specifically mutated in hereditary diseases, a set consisting largely of complex multidomain proteins (Mushegian et al. 1997). Among 84 proteins in the updated disease gene database (XREFdb as of July 1, 1997; see Methods), 68 (81%) shared similarity with nematode proteins, but in the majority of these cases the similarity was limited to individual domains within larger proteins with a different overall domain architecture. By our criteria of orthology, which requires similarity along the entire length of multidomain proteins and not just individual domains (see Methods), 25 human disease proteins (37% of all detected similarities) had candidate orthologs in the sequenced portion of C. elegansproteome (Table 3). Of the 34 human proteins with similarity matches in the fly proteome, 13 proteins (38%) had candidate orthologs in D. melanogaster. Thus, the likelihood of nematode and fly proteins possessing the same domain architecture as human disease gene products was remarkably similar.
Ortholog Conservation in Model Invertebrate Animals
The available portion of the fly proteome contains a high proportion of protein sequences obtained through positional cloning of phenotypically significant genes as well as genes specifically cloned by homology to mammalian proteins. Therefore, one might expect that protein families would be unevenly represented among available Drosophilaproteins. We addressed this issue by querying the Drosophilaand C. elegans proteomes with additional sequences of biological interest. In one query using 30 human enzymes belonging to 4 disparate central metabolic pathways (Table 3), 24 had candidate orthologs in C. elegans and 18 in Drosophila. In another search, using a set of 151 human leukocyte surface (CD) antigens, 78 shared similarity with Drosophila sequences and 89 with C. elegans sequences, although not unexpectedly most of these similarity matches were to portable modules (such as immunoglobulin-like or epidermal growth factor (EGF)-like domains) within a nonorthologous protein. Interestingly, there appear to be three times as many orthologs of human CD antigens in C. elegans as in Drosophila (Table 3). Inspection of the similarixies showed that this difference in the number of orthologs is explained by the almost total absence of certain classes of proteins related to CD antigens among the available Drosophilasequences, including large metalloproteases, the type II (4TM) transmembrane receptors, aminopeptidases, and apyrases.
Concluding Remarks
In this study we evaluated the nematode C. elegans and the fruit fly D. melanogaster as model systems for studying human proteins using protein sequence comparison techniques. By applying strict and reproducible criteria for identifying orthologous proteins, we could extract numerous protein-coding genes for phylogenetic analysis. Our simultaneous analysis of multiple orthologous proteins shows that different proteins can generate different apparent phylogenetic tree topologies, strongly suggesting that historical phylogenies should not be inferred based on a single protein-coding gene. Unequal evolutionary rates are an important factor in calculating phylogenetic trees, and indeed it appears that the majority of C. elegans genes are evolving more rapidly than their Drosophila counterparts. The approaches of ortholog extraction used in this work can be used to better define data sets for phylogenetic analysis among a broader range of representative animal phyla. The available portion of the fly proteome appears to be comparatively enriched in conserved protein domains because of abundant representation of phenotypically defined genes, while missing numerous protein families. The ortholog-to-paralog ratio with regard to human proteins is very similar in the two model animals, indicating that the domain architecture in fly and nematode proteins approximates that of their human homologs to the same extent.
METHODS
Databases
The NR database at the National Center for Biotechnology Information (Bethesda, MD) was used as the source of sequences and for most of the database searches. Species-specific sets of proteins were extracted from NR using Nentrez network tools and the species names as queries. Unfinished genome sequences from the C. elegansgenome project (http://www.sanger.ac.uk/Projects/C_elegans) and database of C. elegans ESTs (http://www.ddbj.nig.ac.jp/c-elegans/html/CE_INDEX.html) were used to verify the protein sequences predicted from genomic DNA. The database of human disease genes and their homologs is available athttp://www.ncbi.nlm.nih.gov/XREFdb, and partial data on orthologs in the nematode and yeast are athttp://www.ncbi.nlm.nih.gov/Disease_Genes. A nonredundant list of human leukocyte surface (CD) antigens was constructed by modifying the list available athttp://www.expasy.ch/cgi-bin/lists?cdlist.txt. Information on biochemical pathways was obtained in part fromhttp://www.genome.ad.jp/kegg.
Sequence Database Searching, Cross-Referencing, and Ortholog Identification
Database searches were performed using the BLAST2 algorithm (Altschul and Gish 1996), with gap width 256 and no filtering. The BLASTP program was used to search protein databases. The TBLASTN program was used to search the nucleotide sequence databases. In all searches, matches with similarity scores <75 were removed. This cutoff eliminates many spurious hits and virtually never eliminates orthologs for medium-sized proteins (A.R. Mushegian, unpubl.). To count the fraction of conserved and unique protein domains, a more restrictive similarity score cutoff, s > 90, was used. BLASTP results were automatically processed using the BLATAX program (Koonin et al. 1996) to extract the best matches in the given species.
Two measures were applied to distinguish candidate orthologs from likely paralogs based on sequence similarity, the BLASTP similarity score and the percentage of amino acid identity in the aligned segments. Criteria used to define candidate orthologs (Tatusov et al. 1996) were as follows. First, protein A in proteome a is a candidate ortholog of protein B in proteome b, if protein B is the best match when sequence A is searched against proteome b, and, conversely, protein A is the best match when sequence B is searched against proteome a. Second, A and B share similarity along their whole lengths. Third, no homolog in a taxonomic outgroup (S. cerevesiae in the present analysis) is closer to A than B, or closer to B than A. Sequences that belong to large, diverged protein families were not considered because of limitations in applying the classic definition of orthology in such cases (Gehring et al. 1994; Tatusov et al. 1997). The following families were thus excluded from analysis: protein kinases, protein phosphatases, RAS-like GTPases and their regulators, chaperones of the HSP60, HSP70, and HSP90 families, and RNA-binding proteins containing RNA recognition motifs.
Phylogenetic Methods
Amino acid sequence data sets for each of 42 protein-coding genes (see Results and Discussion) included orthologs as defined above fromS. cerevisiae, C. elegans, D. melanogaster, and Homo sapiens. Orthologous protein quartets were aligned using a star alignment procedure (Myers and Miller 1988), as implemented in Align Plus software version 3 (Scientific and Educational Software Co.) using the yeast sequence as a guide. This method was chosen because it does not invoke phylogenetic assumptions to carry out the alignment. Each quartet alignment was adjusted interactively using the MACAW program (Schuler et al. 1991) to correct alignment errors, and regions where amino acid similarity were too low to be certain of the alignment were deleted. The alignments are available athttp://chuma.cas.usf.edu/~garey/alignments/alignment.html. Phylogenetic analysis was carried out only with quartet alignments where amino acid identity was >35% among all members. Sequence sites in an alignment with gaps in any single taxon sequence were excluded from phylogenetic analysis. Maximum parsimony trees were produced with the PHYLIP package (Felsenstein 1993). Evolutionary distances and neighbor-joining trees were calculated using both a Poisson distribution of amino acid substitutions and a γ correction (shape parameter = 2) using the MEGA program (Kumar et al. 1994). All trees were tested by the analysis of 100 bootstrap replicates.
Acknowledgments
We thank Carl Johnson for his keen interest in this project and valuable suggestions, Greg Ederer, Maurice Leysens, and Douglas Wood for programming support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.








