
Dispersed Repetitive DNA Has Spread to New Genomes Since Polyploid Formation in Cotton
Abstract
Polyploid formation has played a major role in the evolution of many plant and animal genomes; however, surprisingly little is known regarding the subsequent evolution of DNA sequences that become newly united in a common nucleus. Of particular interest is the repetitive DNA fraction, which accounts for most nuclear DNA in higher plants and animals and which can be remarkably different, even in closely related taxa. In one recently formed polyploid, cotton (Gossypium barbadense L.; AD genome), 83 non-cross-hybridizing DNA clones contain dispersed repeats that are estimated to comprise about 24% of the nuclear DNA. Among these, 64 (77%) are largely restricted to diploid taxa containing the larger A genome and collectively account for about half of the difference in DNA content between Old World (A) and New World (D) diploid ancestors of cultivated AD tetraploid cotton. In tetraploid cotton, FISH analysis showed that some A-genome dispersed repeats appear to have spread to D-genome chromosomes. Such spread may also account for the finding that one, and only one, D-genome diploid cotton, Gossypium gossypioides, contains moderate levels of (otherwise) A-genome-specific repeats in addition to normal levels of D-genome repeats. The discovery of A-genome repeats in G. gossypioides adds genome-wide support to a suggestion previously based on evidence from only a single genetic locus that this species may be either the closest living descendant of the New World cotton ancestor, or an adulterated relic of polyploid formation. Spread of dispersed repeats in the early stages of polyploid formation may provide a tag to identify diploid progenitors of a polyploid. Although most repetitive clones do not correspond to known DNA sequences, 4 correspond to known transposons, most contain internal subrepeats, and at least 12 (including 2 of the possible transposons) hybridize to mRNAs expressed at readily discernible levels in cotton seedlings, implicating transposition as one possible mechanism of spread. Integration of molecular, phylogenetic, and cytogenetic analysis of dispersed repetitive DNA may shed new light on evolution of other polyploid genomes, as well as providing valuable landmarks for many aspects of genome analysis.
[The sequence data described in this paper have been submitted to GenBank under accession nos. AF060571–AF060667 and U31112–U31113.]
Dispersed repetitive DNA is a major component of higher eukaryotic genomes, implicated as a major contributor to variation in DNA content among organisms of similar complexity (Charlesworth et al. 1994). Many dispersed repetitive element families may be examples of selfish DNA (Doolittle and Sapienza 1980; Orgel and Crick 1980) that is free to propagate in genomes unless it impairs the fitness of the organism. Selective advantages conferred by some dispersed repetitive elements have been suggested, such as the recruitment of genes (Martignetti and Brosius 1993), repair of chromosomal breaks (Teng et al. 1996), or induction of favorable mutants (Zeyl et al. 1996).
Dispersed repetitive DNA elements are convenient landmarks for many aspects of genome analysis, such as chromosome walking (Nelson et al. 1989) and transcript isolation (Valdes et al. 1994). Chromosome painting (Liu et al. 1993) by in situ DNA hybridization is an efficient means to identify alien chromatin in hybrid nuclei, providing evidence of rare gene flow in natural populations (Rikke et al. 1995) or introgression of chromosome segments that confer attributes such as disease or pest resistance to crop plants (Jiang et al. 1993, 1994;Nkongolo et al. 1993; Heslop-Harrison and Scharzacher 1996).
The genus Gossypium is a facile system for investigating the genomic organization and evolution of repetitive DNA sequences that become newly united in a common nucleus. Gossypium includes about 50 species, grouped into 7 genome types (A–G) on the basis of chromosome pairing affinities (for review, see Endrizzi et al. 1984). The five polyploid Gossypium species recognized today, including cultivated cottons (G. hirsutum and G. barbadense) are thought to have been spawned about 1–2 million years ago by transoceanic migration of an Old World (A genome) progenitor followed by hybridization with a New World (D genome) progenitor (Wendel 1989). The ancestral A and D genomes are thought to have diverged from a common ancestor about 4–11 million years prior to being reunited in a common polyploid nucleus (Wendel 1989). Extant A- and D-genome cottons share a common chromosome number (n = 13) but exhibit hybrid sterility, and differ by at least nine chromosomal rearrangements (Reinisch et al. 1994), as well as about 0.85 pg (about 45%) in gametic DNA content (H.J. Price and J.S. Johnston, unpubl.).
We have analyzed 83 noncross-hybridizing cotton DNA clones containing dispersed repetitive elements that have been shown previously to comprise about 24% of the tetraploid cotton genome (Zhao et al. 1995). Most dispersed repeat families in tetraploid (AD) cotton are largely restricted to the A-genome diploid ancestors and are absent from most D-genome diploids. In tetraploid cotton, however, some families of these dispersed repeats are found at low levels on chromosomes derived from the D-genome ancestor, suggesting that the repeats have spread since formation of polyploid cotton. Such spread may also account for the finding that one, and only one, D-genome cotton, G. gossypioides, contains moderate levels of the (otherwise) A-genome-specific repeats. The discovery of A-genome repeats in G. gossypioides adds genome-wide support to a suggestion previously based on evidence from only a single genetic locus (Wendel et al. 1995b) that this species may be either the closest living descendant of the New World (D-genome) cotton ancestor, or an adulterated relic of polyploid formation. A likely mechanism for spread of the dispersed repeats appears to be transposition, as chromosomal recombination between the two genomes is rare (if not absent), and the level of divergence between the genomes appears too high for gene conversion. At least four of the repetitive DNA clones show sequence similarity to transposons from other taxa, although a high degree of heterogeneity among the cotton clones is evident. The majority of repetitive DNA clones contain internal inverted or direct repeats, and a subset (including two of the possible transposons) hybridize to mRNAs expressed in the cotton seedling, suggesting that others may also be retrotransposon-like elements. None of these clones corresponds to retrotransposon-like sequences previously reported in cotton (Vanderwiel et al. 1993), suggesting that a very complex population of such elements exists in cotton. Transposition of dispersed repetitive elements might account for rapid genomic restructuring after polyploid formation in other taxa (Song et al. 1995) and/or persistent genetic instability of inbred cotton and other crops. Integrated molecular, phylogenetic, and cytogenetic analysis of dispersed repetitive DNA may shed new light on the ancestry of other polyploids and afford implementation of efficient new techniques for analysis of large genomes such as those of many major crops.
RESULTS
Genome-Specific (or Enriched) Families of Dispersed Repetitive Elements Account for Much of the Difference in DNA Content Between Aand D-Genome Diploid Cottons
We have isolated and identified 83 DNA clones containing non-cross-hybridizing dispersed nuclear repetitive DNA elements fromG. barbadense cultivar Pima S6, a tetraploid cotton containing A and D subgenomes (Zhao et al. 1995). Only a single clone representing each of these 83 families was studied—while individual clones may present a biased representation of a particular family, this sample of 83 clones should accurately represent the population of abundant dispersed repeat families in tetraploid cotton. Noncross-hybridizing clones are tentatively thought to represent different SINE-like (Deininger 1989) repetitive DNA families. It remains a possibility that some clones may represent nonoverlapping fragments from one or more families of LINEs (Smyth 1991); however, each clone was found to detect a unique pattern when hybridized to cotton genomic DNA digested with 13–18 different restriction enzymes (Zhao et al. 1995). Further, the 20 most abundant families each have different FISH karyotypes (Hanson et al. 1998).
The genomic affinity of each clone (Table 1) was evaluated first by hybridization to stoichiometric quantities of DNA from the only two extant A-genome species (G. arboreum andG. herbaceum) and divergent representatives of the D-genome group (G. trilobum and G. raimondii). Clones were classified as genome-specific (>10× difference in signal between A- and D-genome types; Fig. 1A,B), genome-enriched (host genome showed 5–10× difference in signal; see Fig. 1C), or common (similar signal; Fig. 1D). At moderate hybridization stringency (0.5× SSC, 65°C), most clones showed strong signal with both A-genome species and virtually no signal with either D-genome species. Only four clones (5%) showed enrichment or specificity in the D genome (and thus were grouped together).
Dispersed Repeats Account for an Estimated 48% of the Difference in DNA Content between A- and D-Genome Cottons
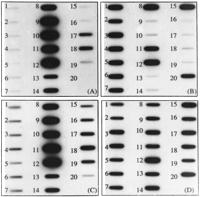
Slot-blot hybridization analysis of genome specificity for cotton dispersed repetitive DNA elements. Slot-blotted genomic DNA from 19Gossypium taxa and an outgroup, hybridized with (A) pXP137, representing an A-genome-specific family; (B) pXP195, representing a D-genome-specific family; (C) pXP224, representing an A-genome-enriched family; (D) pXP215, representing a family common to A and D genomes. Starting with slot 1, genome types and individual taxa are: D genome: (1) G. aridum #123; (2) G. laxum D9-3; (3)G. thurberi D10-9; (4) G. trilobum D8-4; (5) G. klotschianum D3k-55; (6) G. davidsonii #32A; (7) G. raimondii D5-37; (8) G. gossypioides. A genome: (9) G. arboreum, (10) G. herbaceum. AD tetraploid: (11) G. hirsutum Tx9; (12) G. barbadense K101. B genome: (13) G. anomalum;(14) G. triphyllum. C genome: (15) G. robinsonii; (16) G. sturtuanum var. nandewarense AZ40. E genome: (17) G. somalense E2. F genome: (18) G. longicalyx. G genome: (19) G. bickii G1-4. Outgroup: (20) T. lampas. Prior genomic Southern analysis showed that the five tetraploid species are similar in their set of repetitive elements (Zhao et al. 1995); consequently, only two were used here.
Genome-specific and genome-enriched repeat families may account for about half of the difference in DNA content of the A and D genomes (Table 1). Individual DNA clones averaged about 500 bp in length, similar to the typical length of SINEs (Deininger 1989) in other taxa. By multiplying the length of each noncross-hybridizing clone by its previously determined copy number in G. barbadense (Zhao et al. 1995) and summing across classes of genome specificity (A genome, D genome, or common), we estimated that A-genome-specific or enriched clones account for 0.41 pg more DNA than D-genome repeats, or about 48% of the 0.85 pg difference in total DNA content between the A and D genomes. This estimate must be considered only a first-order approximation, contingent on better delineation of the precise boundaries of individual repetitive DNA elements within the clones. Further, because only a single representative of each putative repetitive DNA family was evaluated, our published copy number estimates (Zhao et al. 1995) will tend to underestimate the actual copy number of families in which different elements are highly divergent from each other.
The Phylogenetic Distribution of Dispersed Repetitive Elements Is Generally Consistent with Present Understanding of GossypiumPhylogeny
To investigate further dispersed repetitive DNA evolution inGossypium, we hybridized labeled insert DNA from each family to slot-blotted total genomic DNA of 20 different cotton species, representing each of the 7 recognized genome types and the AD tetraploids (examples in Fig. 1). For each of the 83 repetitive DNA probes applied to slot-blotted DNA of each of the 20 cotton species, signal was quantified by densitometry, standardized relative to the average signal of the two AD-genome species (defined as 1.0), and adjusted for genome size ratios as indicated in Methods to make results directly comparable. Average signal intensities across the 83 probes are plotted for each Gossypium genome type, grouped according to our present understanding of Gossypium phylogeny (Fig.2; Wendel and Albert 1992).
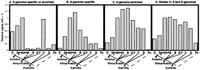
Densitometry analysis of dispersed repetitive DNA hybridization inGossypium. Labeled insert DNA from each of the 83 repetitive DNA clones was hybridized to slot-blotted total genomic DNA of 20 different cotton species, representing each of the 7 recognized genome types and the AD tetraploids (as listed in Fig. 1 legend, except that Dg designates G. gossypioides, and Th. designates the outgroup Thespesia lampas). For each of the 83 repetitive DNA probes applied to slot-blotted DNA of each of the 20 cotton species, signal was quantified by densitometry, standardized relative to the average signal of the two AD-genome species (defined as 1.0), and adjusted for genome size ratios as indicated in Methods to make results directly comparable. Average signal intensities across the 83 probes are plotted for each Gossypium genome type. Gossypiumgenome types are grouped according to our present understanding ofGossypium phylogeny based on chloroplast DNA restriction site variation (Wendel and Albert 1992). Geographic distributions for taxa are also indicated.
With the exception of G. gossypioides (see next paragraph), the distribution of repetitive element families is generally consistent with our present understanding of Gossypium phylogeny (Wendel and Albert 1992). Families that were abundant in the D genome, which is confined to the New World, were rare in the African/Arabian A, B, E, and F genomes (Fig. 2A). A-genome-specific (Fig. 2B) or -enriched (Fig.2C) elements were found at moderate levels in the closely related B, E, and F genomes, at low levels in the Australian C and G genomes, and virtually absent from an outgroup, Thespesia lampas. Common elements (Fig. 2D) were found at similar levels in the A, B, C, E, F, G, and AD genomes, and somewhat lower levels in the small D genome and the outgroup Thespesia.
A few exceptions to this generally congruent picture provide fertile topics for future study. For example, one of the four D-genome families was highly abundant in the C genome of G. robinsonii, and moderately abundant in Thespesia (Figs. 1B and 2A), suggesting convergent amplification of a common ancestral sequence in these taxa.
Among D-Genome Cottons, Only G. gossypioides Contains Dispersed Repeats that Are Otherwise Confined to the A Genome
The set of dispersed repeat families found in G. gossypioides was incongruous with that of any other diploidGossypium genome type. D-genome families occurred at similar levels in G. gossypioides and the other D-genome cottons (Fig.2A). However, signals from A-genome specific repeats were found inG. gossypioides at ∼36% of the level of A-genome diploids and 600% higher than in other D-genome cottons (Fig. 2B). To exclude the possibility that this result was attributable to misidentification of the G. gossypioides accession used, labeled genomic DNA isolated from a second accession of G. gossypioides was applied as a probe to Southern-blotted PCR-amplified insert DNA from each of the 83 repeat families. The G. gossypioides DNA strongly hybridized to these families, while G. raimondii DNA hybridized only to the D-genome-specific families. Moreover, differentG. gossypioides plants were used for slot-blotted DNA and for FISH (see below), but both supported the result (Fig. 3).
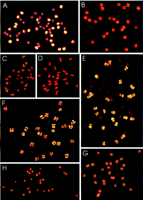
FISH analysis of dispersed repetitive DNAs in cotton. (A–D) Probe pXP224 on G. hirsutum, G. arboreum, G. gossypioides, andG. raimondii, respectively. (E–H) Probe pXP137, onG. hirsutum, G. arboreum, G. gossypioides, and G. raimondii, respectively.
FISH Analysis Reveals Spread of Some Dispersed Repetitive DNA Families in Polyploid Cotton Chromosomes
To evaluate further the physical distribution of dispersed repeats in tetraploid cotton, 20 of the 83 dispersed repeat probes have been applied to the chromosomes of four Gossypium species by FISH (Hanson et al. 1998). Detection of A-genome-specific (or -enriched) families in G. gossypioides, but not G. raimondii,established correspondence of FISH with slot-blot hybridization data (Fig. 1).
Different families of dispersed repeats seem to vary in the extent to which they have spread to a new subgenome. As examples, FISH analysis of two repetitive probes, pXP137 and pXP224, is shown. Both probes hybridize strongly to tetraploid AD cotton (Fig. 3A,E) and to G. arboreum (Fig. 3B,F), an A-genome diploid cotton, consistent with being A genome specific. G. raimondii (Fig. 3D,H), a diploid commonly thought to be the D-genome donor to tetraploid cotton, shows virtually no discernible signal.
The two probes differ markedly in their distribution across the chromosomes of tetraploid cotton. pXP137 (Fig. 3E) clearly distinguishes between the A- and D-subgenome chromosomes of tetraploid cotton, suggesting that it has largely remained confined to the A subgenome. In contrast pXP224 (Fig. 3A) reveals a continuous series of hybridization signals to individual chromosomes, suggesting that it has spread to the D-subgenome chromosomes.
Further evidence in support of the spread of pXP224 comes from the FISH pattern for G. gossypioides (Fig. 3C), which is comparable to the D-subgenome chromosomes of G. hirsutum that fall at the lower end of the continuum of signal intensities. Although pXP137 appears to have remained largely A genome specific in tetraploid cotton, it is present at low levels in G. gossypioides (Fig. 3G).
An extensive survey of FISH karyotypes for many additional families, described in a companion paper (Hanson et al. 1998), suggests that the majority of families resemble the pattern of pXP224, with a continuous distribution of hybridization signal across the chromosomes of tetraploid cotton, suggesting that they have spread to D-subgenome chromosomes.
Most of the Cotton Nuclear Repetitive Elements Do not Correspond to Previously Identified Genes or DNA Sequences
One-pass sequences revealed that 24 (23%) of the 103 repetitive DNA clones (including both tandem and dispersed repeats) showed significant correspondence (BLAST>150) to previously identified DNA sequences from a wide range of organisms (Table 2).
Correspondence of Cotton Nuclear Repetitive Elements to Other Organisms’ Genes or DNA Sequences
Three of the clones show correspondence to parts of a transposable element from Lilium (Fig. 4). pXP030 and pXP1-58 are highly divergent from one another but each shows a high degree of DNA sequence similarity to partially overlapping regions of a transposon discovered in Lilium henryi (Smyth et al. 1989) that also corresponds to elements found in Nicotiana (Royo et al. 1996), Brassica (GenBank accession no. X99804), andArabidopsis (GenBank accession no. Z97342). pXP067 does not overlap with either pXP030 and pXP1-58, but does correspond to a different region of the same Lilium element. The regions of correspondence between the Lilium element and each of the three cotton elements include portions of the L. henryiintegrase region (Fig. 5). A fourth cotton element, pXP1-13 corresponds to part of a Drosophila melanogastertransposon (Biessmann et al. 1992).
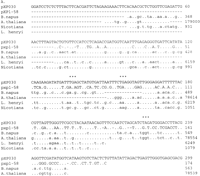

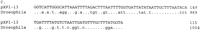
DNA sequence alignments of cotton repetitive elements, pXP030, pXP1-58, pXP067 and pXP1-13 (uppercase letters) with putative retrotransposon sequences from other organisms (lowercase letters). Identical bases are indicated by dots. Sequences without matches or gaps are indicated by dashes. Stop codons are indicated by asterisks. (A) pXP030 and pXP1-58 are multi-aligned with a Brassica napus DNA fragment containing a retrotransposon integrase motif (GenBank accession no.X99804); Arabidopsis thaliana DNA chromosome 4, ESSA I contig (GenBank accession no. Z97342); Nicotiana alataretrotransposon Tna1-2 integrase motif (Royo et al. 1996), and part of the downstream sequence of a L. henryi del transposon (Smyth et al. 1989). (B) pXP067 encodes a region that resembles part of the upstream sequence of a L. henryi del retrotransposon (Smyth et al. 1989). (C) pXP1-13 contains a sequence with similarity to D. melanogaster transposable element HeT-A-RT394 (Biessmann et al. 1992).

Reduced amino acid sequence alignments of pXP030, pXP1-58, and pXP067 with L. henryi integrase region. Identical amino acids are indicated by dots above the amino acid sequence. (Asterisks) Stop codon positions; (dashed line) sequence gap. Frameshifts are also shown by indication of the frame that has homology with the subject sequence.
The vast majority of cotton nuclear repetitive elements show a high degree of internal repetition. A total of 89%, 81%, and 59% of the high-abundance, middle-abundance, and low-abundance repetitive DNA clones, respectively, showed internal subrepeats of >20 nucleotides in length. Only four of the internal repeats were perfect duplications—three in direct orientation and one in inverse orientation (see examples in Fig. 6). Among the 95% of subrepeats that were imperfect (differing by no more than 3 consecutive nucleotides), 57% were in inverse orientation and 38% in direct orientation.
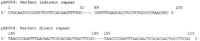
Examples of internal subrepeats within repetitive DNA clones. (Dashes) Spaces between two repeat units. The numbers above each repeat unit are nucleotide positions in the DNA sequence of the indicated repetitive DNA clone.
Some of the Repetitive DNA Clones Hybridize to Transcripts
Hybridization of radioactively labeled first-strand cDNA made from seedling poly(A)+ RNA, to slot-blotted DNA from the individual repetitive clones (prepared as described by Zhao et al. 1995) showed that a subset hybridize to mRNAs that are expressed in young cotton seedlings. Replica slot-blots were hybridized to labeled poly(A)+RNA and to labeled plasmid DNA from the cloning vector. The relative signal levels (poly(A)+ RNA/plasmid) are shown in Fig.7. Most clones showed relative signals that were similar to that of the negative control (slot-blotted plasmid DNA, ratio = 0.09), but 12 of the clones showed relative signal of >10, more than 100 times greater than the negative control. (Fig.7). Among the 12 clones hybridizing to mRNAs were pXP067 and pXP1-58, two of the three clones resembling the Lilium transposon, and clones that have DNA sequences similar to the Arabidopsiscyc2b gene, and Plasmodium DNA polymerase α gene.
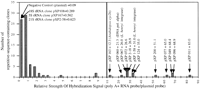
Hybridization of radioactively labeled first-strand cDNA made from seedling poly(A)+ RNA to 1 μg of slot-blotted DNA from the individual repetitive clones. Hybridization intensity is expressed as a ratio of signal (quantified by densitometry) from hybridization with labeled cDNA to signal from hybridization with labeled plasmid DNA. Triplicate values for the negative control (1 μg of slot-blotted plasmid DNA) averaged 0.09. A total of 12 individual repetitive DNA clones showed ratios of >10, or 100 times the negative control. DNA sequences corresponding to high-signal repetitive DNA clones are indicated.
Three repetitive clones that were not among those hybridizing to mRNAs (pXP2-38, pXP069, pXP101) had poly(A) signals (5′-AATAAA-3′), possibly suggesting that these clones include the 3′-untranscribed regions of coding sequences.
DISCUSSION
Dispersed repetitive DNA families, which account for much of the difference in DNA content among diploid cotton species, appear to spread readily to new genomes. About 70% of angiosperm genomes are thought to be recent or ancient polyploids (Stebbins 1966; Masterson 1994), as well as many vertebrates (Atkin and Ohno 1967; Hinegardner 1968; Ohno et al. 1968; Nadeau and Sankoff 1997). Many evolutionary models suggest that polyploid formation should be associated with a selective advantage, favoring divergence of the parental genomes, to facilitate the bivalent chromosome pairing observed in derived polyploids such as cotton (Kimber 1961). However, in contrast, genome-specific dispersed repeat families have become more uniformly distributed across the cotton subgenomes following polyploidization, spreading to the genome that did not previously contain them. These data contraindicate a direct role of dispersed repetitive DNA in regulating chromosome pairing and favor alternative mechanisms based on chromosome structure (Rieseberg et al. 1995; C.L. Brubaker, A.H. Paterson, J.F. Wendel, in prep.) and/or specific genes (Riley and Chapman 1958).
G. gossypioides: Progenitor or Relic?
Tracing the ancestry of polyploids is often difficult because of heterogeneity within and among candidate donor taxa and ongoing evolution of these taxa subsequent to polyploid formation (sometimes in competition with the new polyploid). The observation that dispersed repeats can spread to new genomes, suggests that ancient genetic exchange between divergent populations may have left molecular footprints of dispersed repetitive DNA.
Its set of dispersed repeat families provides genome-wide support for the proposal that G. gossypioides, rather than its sisterG. raimondii, may be the closest living descendant of the New World (D-genome) cotton ancestor (Reinisch et al. 1994). The interpretation that G. gossypioides is a D-genome cotton is compelling, supported by geographic distribution, cytogenetic data, which show that its chromosome morphology and DNA content are similar to those of other D-genome taxa (Brown and Menzel 1952; Endrizzi et al 1984), crossing experiments, which show that G. gossypioidesforms fertile hybrids only with G. raimondii (Menzel and Brown 1955), and phylogenetic analysis of chloroplast DNA restriction sites, which places G. gossypioides deeply nested within the D-genome clade, sister to G. raimondii (Wendel and Albert 1992).
On the basis of slot-blot hybridization signals (Figs. 1,2), about 400,000 copies of (otherwise) A-genome-specific dispersed repeats are widely distributed throughout the genome of G. gossypioides. Formation of an interspecific hybrid that combined the A and D genomes in a common nucleus may have first permitted A-genome repeats to spread to the D genome. The finding that all tetraploid cotton taxa are indigenous to the New World, but have an A-genome (Old World) cytoplasm, shows that such a hybrid did exist, even if only briefly (Wendel 1989). Diploid A × D hybrids between extant taxa are sterile; however, backcrosses to native D-genome plants might have spawned present-day G. gossypioides, whereas polyploid formation by any of several possible paths (Harlan and deWet 1975) permitted recurring colonization of the D subgenome. Any rare events that conferred selective advantages (Martignetti and Brosius 1993; Teng et al. 1996; Zeyl et al. 1996) might have contributed to divergence of G. gossypioides and G. raimondii. The inability of G. gossypioides to form fertile hybrids with D-genome taxa other than G. raimondii may have helped to contain the further spread of A-genome repeats.
The addition of 400,000 dispersed repeats to the G. gossypioides genome would increase its DNA content by about 0.1 pg (on the basis of the size estimates we used above). While published data suggest that the DNA content of G. gossypioides is similar to that of other D-genome cottons (Edwards and Endrizzi 1976;Bennett and Leitch 1995; H.J. Paterson and S. Johnston, unpubl.), the range among these estimates exceeds the small 0.1-pg difference in DNA content that might be attributable to A-genome repeats (see Table 1footnote).
Possible Mechanisms by which Dispersed Repeats Might Spread to New Genomes
The spread of dispersed repeats to new genomes that appear to lack homologous elements impels consideration of different mechanisms from those that may account for concerted evolution of tandemly repeated elements such as nor (rDNA; Wendel et al. 1995a,b).
Replicative transposition is clearly implicated as one possible mechanism by which at least some of the repeat families may have spread that would explain the spread of A-genome repeats to both G. gossypioides and to the D subgenome of tetraploid cotton. Moderately repetitive families of retrotransposon-like elements have been reported previously in cotton (Vanderwiel et al. 1993), and four of the repetitive DNA-containing clones found in this study correspond to DNA sequences of two known transposons. At least two of these clones, both corresponding to the L. henryi integrase region, hybridized to transcripts that are expressed at readily discernible levels in cotton seedlings. Three additional clones contained poly(A) signals, suggesting that they may derive from the 3′-untranslated region of a coding sequence. The vast majority of clones showed internal subrepeats, in either direct or inverse orientation, a feature commonly associated with transposable DNA sequences. The high tendency for clustering of individual family members (Figure8) implicates a propagation mechanism that has a proximal bias, such as that observed for many transposons. It is noteworthy that none of the retrotransposon-like clones found in this study showed close correspondence to those reported previously in cotton (Vanderwiel et al. 1993), or to each other (see above), suggesting that a very complex population of such elements exists in cotton.
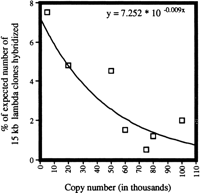
Dispersed repetitive DNA family members tend to be clustered in cotton. About 5000 plaques from a λ phage library of G. hirsutum(average insert size, 15 kb) were hybridized with each of seven different probes containing dispersed repeats. The seven families ranged from about 4,000 to 100,000 copies, spanning the full range of copy numbers for dispersed repeats discovered in cotton. The actual number of recombinant phages containing each repeat family was compared to the number that would be expected if individual family members were evenly distributed throughout the cotton genome. Each of the seven repeat families shows a high degree of clustering, as they are found on far fewer λ clones than would be expected if individual family members were evenly distributed throughout the genome. High-abundance families were found on as few as 1%–2% of the expected number of λ clones, a much greater degree of clustering than moderate-abundance families.
A possible alternative mechanism of spread, gene conversion, is well documented in yeast (Ernst et al. 1981; later examples reviewed inPetes et al. 1988) and mammals (Murti et al. 1994). However, gene conversion appears to require at least 134 bp of perfect, uninterrupted homology between donor and recipient sites (Waldman and Liskay 1988). Abundant RFLP variation between A- and D-genome diploid cottons suggests that such highly conserved sites may be rare (Reinisch et al. 1994; C.L. Brubaker, A.H. Paterson, and J.F. Wender, in prep.).
Other mechanisms seem unlikely to account for the spread of genome-specific repeats to new genomes in cotton. Pairing and recombination between homologous chromosomes in different subgenomes does not appear to have substantially (if at all) affected the organization of the modern cotton genome (Reinisch et al. 1994). Further, this mechanism does not readily account for colonization ofG. gossypioides, as diploid hybrids between extant A- and D-genome cottons are sterile. [Polyploid formation presumably involved unreduced gametes or some other mechanism, leading to formation of a tetraploid that could enjoy normal bivalent pairing, (Harlan and deWet 1975)]. Finally, the genome-specific distribution of some elements such as pXP137, is inconsistent with the occurrence of homologous exchange.
A question for further analysis is the extent to which the spread of genome-specific repeats is a general property of polyploid taxa. The reuniting of two genomes in a common nucleus after 4–11 million years of divergence (Wendel 1989) might create the sort of genome stress (McClintock 1984) that could precipitate punctuational evolution.
Applications of Dispersed Repeats to Analysis of Large Genomes
The utility of dispersed repetitive DNA in genome analysis is well established in mammals (briefly reviewed above), but has been much less widely exploited in plants. The cotton dispersed repeats provide the means to quickly establish large numbers of landmarks throughout the genome useful for integrating genetic and physical maps, fingerprinting individual BAC clones, and as sequence-tagged sites for future genomic sequencing.
One impetus for our work was to develop tags to determine whether particular BACs from tetraploid cotton were from the A subgenome or the D subgenome to expedite chromosome walking to agriculturally important alleles that mapped to one specific subgenome of tetrapolid cotton (most of the world’s cultivated cotton is tetraploid). The ∼15%–20% of nonhomogenizing interspersed repetitive element (IRE) families that remain largely restricted to their source genome in tetraploid cotton (Hanson et al. 1998), collectively provide about 280,000 such tags.
A high degree of clustering observed within individual repeat families may impede some applications to plant genome analysis, such as BAC tagging. To tag an A-subgenome-derived BAC clone of 100 kb, at least 7% (18,000) of individual family members would have to be distributed at average intervals of 100 kb along the ∼1800 Mb of the A subgenome. By screening smaller (15 kb) λ clones, we found a high degree of clustering among family members, with an individual family tagging an average of only 0.7% of λ clones. This clustering was consistent with the strong concentrations of signal found by FISH analysis of most repetitive DNA probes. Clustering of repeat units further supports the possibility that transposition, with a proximal bias, was an important propagation mechanism (see above). Such clustering has also been found in other large-genome plants, where most repetitive elements in the cluster were shown to have transposed (San Miguel et al. 1996). Moderate-abundance families appear much more widely dispersed than high-abundance families (Fig. 8)—pools of moderate-abundance families may be more suitable than individual high-abundance families as probes for comprehensive fingerprinting of large DNA clones (Nelson et al. 1989).
Better understanding of cotton dispersed repeat families might yield practical strategies for insertional mutagenesis, analogous to those employed for multiple-copy transposons (Chomet 1994). In total, the A-genome dispersed repeat families are estimated to include about 1.4 million individual elements, a potentially powerful mutagen even if only a small subset is still capable of spread. Naturally occurring insertional mutagenesis might partly account for the widespread observation that inbred cottons remain far more variable in phenotype than would be expected after many generations of selfing. Further, such insertional mutations might account for new RFLP alleles found subsequent to polyploid formation in other taxa such asBrassica (Song et al. 1995), a mechanism that would be consistent with the tendency for both Brassica and cotton (Reinisch et al. 1994) RFLPs to be revealed by multiple restriction enzymes.
Comprehensive isolation and analysis of dispersed repeat families may provide valuable new information about the genomes of other polyploid plants, such as soybean, wheat, oats, tobacco, and canola. Only small numbers of dispersed repeat families have been characterized in most of these taxa (Smyth 1991; Anamthawat and Heslop-Harrison 1993). By comprehensive cloning of the major dispersed repeat families in a taxon, one isolates the genomic DNA that accounts for most variation in genome size. This nemesis to chromosome walking, once cloned and characterized, becomes a powerful tool for genetic and evolutionary studies.
METHODS
Germ plasm used in this study (Fig. 1, legend) was provided by sources cited previously (Zhao et al. 1995). A second accession ofG. gossypioides, used to confirm the slot blots as described, was generously provided by E. Percival (USDA-ARS, College Station, TX).
Identification and cloning of repetitive DNA elements and estimation of copy numbers were as described (Zhao et al. 1989, 1995). Genome specificity for repetitive element families was evaluated first by Southern analysis of equal genome equivalents of DNA from G. arboreum, G. herbaceum (A genome; 3 μg of DNA), G. trilobum, and G. raimondii (D genome; 2 μg of DNA). Subsequently, individual elements were hybridized to replica slot blots of 0.5 μg of DNA from each of 17 diploid Gossypium species and T. lampas, and to 1 μg of DNA from two tetraploids (listed in Fig. 2, legend), as described (Zhao et al. 1989). Relative signal intensities (Fig. 2) were adjusted for genome size by use of multipliers of 0.35 for D genome and 0.65 for A, E, F, and B genomes on the basis of direct measurement (H.J. Paterson and S. Johnston, unpubl.), and 0.8 for C genome and 0.7 for the G genome on the basis of ratios of DNA content in the respective diploids to that of the tetraploid (Edwards and Endrizzi 1976). Each replica slot blot was used only once.
Selected dispersed repeat families were investigated by FISH on metaphase chromosomes of G. arboreum, G. hirsutum, G. raimondii, andG. gossypioides, as described (Hanson et al. 1996).
Evaluation of clustering used a lambda-Dash II genomic library ofG. hirsutum, prepared and screened according to the manufacturer’s instructions (Stratagene).
Acknowledgments
This research was supported by the US Department of Agriculture, Texas Higher Education Coordinating Board (A.H.P., D.M.S.), National Science Foundation (J.F.W.), and Texas Agricultural Experiment Station.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 These two authors contributed equally to this paper.
-
↵4 Present address: University of Michigan Medical Center, MSRB-II, C568, Ann Arbor, Michigan 48109-0672 USA.
-
↵5 Corresponding author.
-
E-MAIL ahp0918{at}acs.tamu.edu; FAX (409) 845-0456.
-
- Received September 5, 1997.
- Accepted April 2, 1998.
- Cold Spring Harbor Laboratory Press








