
Simultaneous Genotyping and Species Identification Using Hybridization Pattern Recognition Analysis of Generic Mycobacterium DNA Arrays
- Thomas R. Gingeras1,7,
- Ghassan Ghandour1,
- Eugene Wang1,
- Anthony Berno1,
- Peter M. Small2,
- Francis Drobniewski3,
- David Alland4,
- Edward Desmond5,
- Mark Holodniy6, and
- Jorg Drenkow1
- 1Affymetrix, Santa Clara, California 95051 USA; 2Division of Infectious Disease, Stanford University, Stanford, California 94305 USA; 3Public Health Laboratory and Medical Microbiology, Kings College School of Medicine and Dentistry, East Dulwich Grove, London SE22 8QF, UK; 4Division of Infectious Disease, Montefiore Medical Center, Bronx, New York 10467 USA; 5Microbial Disease Lab, California Department of Health, Berkeley, California 94704 USA; 6Palo Alto VA Medical Center, Palo Alto, California 94304 USA
Abstract
High-density oligonucleotide arrays can be used to rapidly examine large amounts of DNA sequence in a high throughput manner. An array designed to determine the specific nucleotide sequence of 705 bp of therpoB gene of Mycobacterium tuberculosis accurately detected rifampin resistance associated with mutations of 44 clinical isolates of M. tuberculosis. The nucleotide sequence diversity in 121 Mycobacterial isolates (comprised of 10 species) was examined by both conventional dideoxynucleotide sequencing of the rpoB and 16S genes and by analysis of the rpoB oligonucleotide array hybridization patterns. Species identification for each of the isolates was similar irrespective of whether 16S sequence, rpoBsequence, or the pattern of rpoB hybridization was used. However, for several species, the number of alleles in the 16S andrpoB gene sequences provided discordant estimates of the genetic diversity within a species. In addition to confirming the array’s intended utility for sequencing the region of M. tuberculosis that confers rifampin resistance, this work demonstrates that this array can identify the species of nontuberculous Mycobacteria. This demonstrates the general point that DNA microarrays that sequence important genomic regions (such as drug resistance or pathogenicity islands) can simultaneously identify species and provide some insight into the organism’s population structure.
[The sequence data described in this paper have been submitted to GenBank under accession nos. AF09766–AF059853 and AF060279–AF060367.]
For patients infected with Mycobacteria, especially those coinfected with the human immunodeficiency virus type 1 and type 2 (HIV-1, HIV-2), the identity of the Mycobacterium species and the presence of mutations that confer both biologically and clinically important phenotypes are of critical importance. Both of these issues have implications for the appropriate care and treatment of the infected patient. For example, although M. avium complex (MAC) is the most common cause for both disseminatedMycobacterium disease and death in patients with AIDS in the developed world (∼25%–50% of adults and 10% of children with AIDS are infected [Inderlied et al. 1993], Mycobacterium tuberculosis infections are also found in these patient populations. Important public health and patient management decisions (e.g., the need for clinical isolation and the choice of the appropriate therapeutic regimen) depend on a timely and accurate identification of the infecting agent. Additionally, almost 10% of newM. tuberculosis patients in the United States show resistance to at least one of the first line antituberculosis drugs (isoniazid [INH], pyrazinamide [PZA], rifampin [RIF], ethambutol [EMB], and streptomycin [STR] with ∼2%–3% of cases resistant to both INH and RIF (Moore et al. 1997).
On the basis of the insights provided by previously characterized RIF resistant mutants in Escherichia coli (Ovchinnikov et al. 1983; Jin and Gross 1988), mutations in the β-subunit of the RNA polymerase (rpoB gene) of M. tuberculosis were first identified and characterized by Telenti et al. (1993). Approximately 90%–95% of RIF-resistant M. tuberculosis strains have been found to possess mutations in an 81-bp section (Musser 1995) of the 3534-bp coding region of the rpoB gene (Miller et al. 1994). Interestingly, of the 122 M. tuberculosis isolates analyzed byTelenti et al. (1993), no polymorphisms other than those conferring drug resistance were observed in 411 nucleotides analyzed in each sample. In this study, we have explored the sequence diversity in a larger segment of the rpoB gene for 10 species of theMycobacterium genus. By use of a high-density oligonucleotide array to derive both hybridization patterns and nucleotide sequences, information from a conserved 705-bp region of the rpoB gene permitted the simultaneous species identification speciation of 121 isolates from 10 Mycobacterium species as well as the detection of mutations that confer RIF resistance in 41 M. tuberculosis isolates.
Genotypic analyses of the Mycobacterium species isolates (Table 1) used in this study were performed with a high-density oligonucleotide array (DNA chip) with probes complementary to the M. tuberculosis rpoB gene sequence. The array served as a generic genotyping chip that provided both specific nucleotide sequence as well as patterns of hybridization highly specific for each species. This demonstrates the general capability of such an array to provide important clinically relevant and biological information about the rpoB genes of related Mycobacterial organisms that have not been sequenced previously.
Mycobacterium Isolates
RESULTS
Rifampin-Conferring Mutations in the rpoB Gene ofM. tuberculosis
A total of 705 of the 3534 nucleotides of the M. tuberculosis rpoB gene was analyzed by use of a high-density oligonucleotide array (Fig. 1). Although this segment of therpoB gene has a GC content of 67.7%, it was resequenced by use of the array with 100% concordance with dideoxynucleotide methodology. A collection of 63 M. tuberculosis isolates gathered from New York and San Francisco areas was analyzed by theM. tuberculosis rpoB array for mutations that confer rifampin resistance. The resistance to rifampin for 44/63 samples was determined prior to the genotypic analyses by collaborating laboratories and the results kept confidential from us until completion of genotypic analyses. Of the 44 M. tuberculosis isolates that were phenotypically resistant to rifampin, 40 possessed mutations associated previously with resistance. One additional isolate (TB40) displayed a mutation (Gln-513 Glu) not described previously. Each array-derived nucleotide sequence was confirmed by use of conventional dideoxynucleotide sequencing. Mutations at codons 531, 526, and 513 (E. coli codon numbering system) were observed to occur most frequently in the rifampin resistant isolates at 35%, 28%, and 25%, respectively (Fig. 1). Mutations were not observed in any of the 705 nucleotides analyzed of the rpoB gene for three of the phenotypically resistant isolates by use of either array or dideoxynucleotide sequencing methodologies.
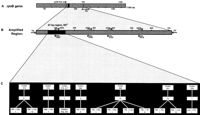
(A) Analysis of a 705-bp region (codons 482–715, E. coli numbering) of the Mycobacterium rpoB gene (1164 amino acids). (B) Region encompassing the 81-bp domain, described previously as having all of the mutations that correlate with the decreased sensitivity to rifampin (Musser 1995). Arrows indicate locations of PCR amplification primers and all of the primers used for dideoxynucleotide sequencing (see Methods for sequence). (C) Codon positions within the 81-bp region, containing mutations in 41 of the 44 RIF-resistant M. tuberculosis mutants. The frequency and type of mutation observed at each codon is presented.
Allelic Frequency and Species-Specific Polymorphisms Present inrpoB and the 16S Genes of Mycobacterium
The rpoB and 16S genes from nine species ofMycobacterium were analyzed at the nucleotide level by use of dideoxynucleotide-based methodology and compared with the M. tuberculosis sequence of these genes. A total of 83 and 82Mycobacterium isolates were characterized for both therpoB and 16S genes, respectively (Table 2). In comparison with M. tuberculosis, an average of 80 polymorphic positions were observed within the 705 nucleotides ofrpoB for each species (Table 2A) and on the average, 21 polymorphic positions were seen within the 180 nucleotides of the 16S gene (Table 2B). Several of the polymorphic positions were observed to be species specific, with a subset of these present in every isolate of that species (conserved polymorphism). For each of 63 M. tuberculosisisolates, no other polymorphic sequences were observed in the rpoB+ene, except for mutations conferring resistance to rifampin.
Analysis of Polymorphisms in the rpoB and 16S Genes of Mycobacterium
Interspecies variation for the 705 nucleotide region of therpoB gene ranged from 14.3% (Micobacterium chelonaeand M. xenopi) to 4.1% (M. avium and M. scrofulaceum) (Fig. 2). Intraspecies variation for rpoB was highest for M. smegmatis (4.2%) andM. gordonae (3.7%) with M. tuberculosis and M. xenopi exhibiting no nucleotide variation in >70 isolates analyzed. For both genes, M. kansasii and M. intracellulare isolates displayed only one-fifth to one-third as many alleles as isolates examined, whereas M. smegmatis andM. fortuitum displayed as many alleles as isolates examined (Table 3). Interestingly, a contrasting view of the diversity within a species group was observed depending on whether therpoB or 16S genes were examined. For example, for M. xenopi, one and three alleles for rpoB and 16S genes, respectively, were observed. In contrast, M. intracellulareexhibited one and four alleles for the 16S and rpoB genes, respectively. Finally, M. tuberculosis and M. fortuitum were similar in complexity of their species groups as exemplified in the number of alleles observed for the 16S and rpoB Genes.
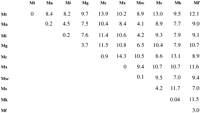
Interspecies and intraspecies variation observed a 10 species ofMycobacterium. Values represented are percent variation based on 705 bp of the rpoB gene for M. tuberculosis (Mt),M. avium (Ma), M. intracellulare (Mi), M. gordonae (Mg), M. chelonae (Mc), M. xenopi (Mx),M. scrofulaceum (Msc), M. smegmatis (Ms), M. kansasii (Mk), M. fortuitum (Mf). Data from M. scrofulaceum are represented by two isolates.
Allelic Variations in MycobacteriumSpecies using 16S and rpoBGenes
Species Identification Based on DNA Sequences of 16S and rpoBGenes
DNA sequence analysis of 705 bases of the rpoB and 180 bases of the 16S genes for 81 of the 121 Mycobacteriumisolates (Table 1) was determined by use of conventional dideoxynucleotide methodology. Analyses of these sequences permitted the clustering of each of the isolates into groups on the basis of either the 16S or rpoB sequences (Fig. 3). The confidence values for each species clusters indicated the groupings were very stable. The bootstrap values ranged from 100% to 69.3% for 16S gene sequences and from 100% to 71% for the rpoB gene sequences. The lowest confidence values were observed in clusteringM. fortuitum and M. scrofulaceum isolates with 16S and rpoB sequences, respectively. Of the 81 isolates, 75 were grouped into the same species clusters by use of either gene sequences. Five of the 81 isolates (m4, m36, m48, m66, m125) were assigned to different species clusters, depending on whether the 16S orrpoB sequence was used as the basis for analysis. Specifically, species identification on the basis of the rpoBsequence for three of the five isolates (m125, m48, m4), was in agreement with that assigned by standard microbiological methods used by the providing laboratories, but differed in assignment on the basis of 16S sequence analysis. Two of the five isolates (m36, m66) were placed in one of three different species clusters depending on the gene sequence analyzed or the microbiological assay used.
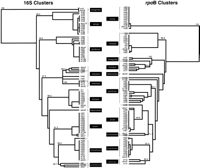
Representation of clusterings of Mycobacterium isolates among 10 species of Mycobacterium. The tree is unrooted and is based on the nearest neighbor joining method for the 180- and 705-bp DNA sequences of the 16S and rpoB genes, respectively, from each of the isolates. Stability of the clusters were evaluated by use of 1000 cycles of bootstrapping (values on tree branches). The six isolates that are assigned to different clusters, depending on the gene sequence used, are highlighted in gray (see text for discussion).
Analyses of both 16S and rpoB sequences together were useful in providing more precise species identification or clarification for 10 of the 81 isolates. Of these 10 isolates, 7 (m91, m104, m68, m64, m65, m67, m71) were identified as MAC or avium–intracellulareby the laboratories of origin. On the basis of both 16S orrpoB sequence analyses, six of the seven isolates were unambiguously identified as M. avium, with m68 identified asM. xenopi. The remaining three isolates m77, m27, and m28 were identified by microbiological assays as M. smegmatis, M. avium, and M. avium, respectively. Analysis with both gene sequences clustered these isolates as M. fortuitum (m77), M. intracellulare (m27), and M. intracellulare (m28), respectively.
Species Identification Using Hybridization Pattern Recognition Analysis of High-Density Oligonucleotide Arrays
The high-density oligonucleotide array used to detect the mutations conferring rifampin resistance in M. tuberculosiswas also used to simultaneously genotype and speciate nontuberculous isolates. As the interspecies sequence variation for the rpoBgene of some of the nontuberculous isolates was >10% (Fig. 2), significant portions of the hybridization patterns produced from nontuberculous rpoB amplicons were unique (Fig.4A). Each hybridization pattern can be represented as a plot of the fluorescence intensities as a function of the base position in the sequence of rpoB (Fig. 4B). When nontuberculous DNA amplicons were hybridized, the fluorescence intensities of the many of the interrogating probes were reduced within the regions of the rpoB sequences (Fig. 4A). This reduction in hybridization intensity affected the ability to determine specific nucleotides, because allele-specific probes for some of the interrogated bases do not exist on the array (Fig. 4C; Table4). However, even though the arrays were designed for the M. tuberculosis gene sequence, the sequence of most of the polymorphic positions for each species could be determined (see legend to Fig. 4C). Repeated measurements indicated that highly specific and reproducible hybridization patterns characteristic of each of the Mycobacterial species could be obtained.

(A) Hybridization patterns produced on an oligonucleotide array that have probes selected to be complementary to the 705 bases ofM. tuberculosis rpoB gene sequence. The amplified, fluorescently labeled 705-bp antisense product from the rpoBgenes of M. tuberculosis and M. gordonae are presented. A total of 5648 oligonucleotide probes were used to interrogate each of the 705 bp in the amplified product. (B) The intensity of hybridization for each of the 705 probes that are complementary to the wild-type sequence (Miller et al. 1993) of theM. tuberculosis rpoB gene is plotted as a function of the base interrogated in the gene sequence. The blue and red plots are the intensity profiles of M. tuberculosis and M. gordonaeimages shown in A. The intensities are obtained from GeneChip software (see Chee et al. 1996; Kozal et al. 1996) and are plotted and compared using the Ulysses software program (Chee et al. 1996). (C) The identity of each base in the 705 bp of therpoB amplicons is determined by the hybridization results of eight probes (four for each strand). The sequences derived from the images in A for M. tuberculosis (M-tb) and M. gordonae (M-go) are shown. Differences between the two genes are denoted by highlighted bases in the M. gordonae sequence. Of these differences, 61% (95/155) of the positions can be identified as specific polymorphic differences between the two species. The remainder of the differences are unidentified or marked by IUPAC ambiguity code. Of the positions identified as a polymorphic difference 7/33 bases correspond to species-specific polymorphisms present in all isolates ofM. gordonae (Table 2).
rpoB Genotyping of Nontuberculosis Isolates with M. tuberculosisArray
The hybridization patterns produced on the M. tuberculosis rpoB arrays for each of 121 Mycobacterium isolates were analyzed by use of two algorithmic approaches to carry out species identification. The first algorithm used a straightforward linear regression analysis of the 1410 (705 bases on each strand) intensities for the probes discovered to be complementary to the wild-type sequence of the M. tuberculosis rpoB gene. This approach selectively analyzed each of the Mycobacterium isolate sequences with only 25% of the probes present in the array. The results of this clustering were represented in a color contour plot (Fig. 5A). The color of each pairwise comparison of the 1 − r 2 values correlated to highly correlated (1-blue) to uncorrelated (0-red) pairs. Interestingly, isolates m125, m66, and m4, which, on the basis of their DNA sequences, were observed to group outside of their biochemical/microbiologically defined species designations, are also observed to cluster outside their biochemically defined groups by this method. Most notably, all of the other isolates were clustered into the same species groups as predicted by the 16S and rpoB sequences.
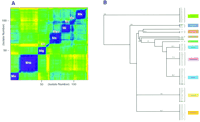
(A) Results of hybridization patterns of 121Mycobacterium isolates analyzed by linear regression assays (SAS Institute 1990). Only probes complementary to the wild-type sequence of the rpoB gene of M. tuberculosis were used in the analysis (i.e., 25%) of probes. The pairwise comparison of the 1 − r 2 values for each of the 121 isolates was performed and like values clustered. The contour plot represents values for 1 (purple) to 0 (red). (Mx) M. xenopi, (Mt) M. tuberculosis, (Mg) M. gordonae, (Mf) M. fortuitum, (Msc) M. scrofulaceum, (Mi) M. intracellulare, (Mc) M. chelonae, (Ms) M. smegmatis, and (Mk) M. kansasii. (B) Results of hybridization patterns analyzed by use of principle component assays. Using all of the 5648 probes on the rpoB array, each of the isolates was clustered on the basis of 15 orthogonal components. The clustering of the isolates was represented by an unrooted nearest neighbor joining tree. The six isolates noted in Fig. 3 are highlighted in this tree.
The second algorithmic approach used to analyze individual hybridization patterns was based on principal component analysis of all of the 5640 probes (705 bases × 2 strands × 4 probes/interrogated base) on the rpoB array. Unlike the earlier linear regression analysis, in this approach, no prioritization for the M. tuberculosis derived, perfect match probes were used. The most informative probes were identified by variance-based variable reduction followed by the principal component analysis of the covariance matrix and reduced the total probe set 15 orthogonal components. These components accounted for 93% of the observed variability in the probe intensities. By use of the same hierarchical clustering procedure used to group the isolates on the basis of linear regression correlation coefficients, a single linkage clustering result is displayed as a hierarchical tree structure (Fig. 5B). Confidence values again showed the clusters to be highly stable. The values ranged from 100% (M. smegmatis, M. xenopi, M. kansasii) to 77.6% (M. intracellulare). The membership in each of the major species clusters were identical to those clusters derived from therpoB sequences with the exception of some of the six isolates highlighted in the DNA clusterings (Fig. 3). Of the five isolates that clustered in different species groups, depending on the gene sequence used to carry out the cluster analysis (m4, m36, m48, m66, m125), only samples m36 and m125 were not grouped as they were by theirrpoB DNA sequence.
DISCUSSION
Hybridization of DNA to high-density oligonucleotide arrays offers the possibility of examining large amounts of sequence with a single hybridization step. The utility of this approach was recently demonstrated by the complete analysis of the entire human mitochondrial genome (Chee et al. 1996). Other applications of these arrays have included the sequence analysis of viral (Kozal et al. 1996) and human genomic sequences (Hacia et al. 1996), quantitative measurements of multiple murine (Lockhart et al. 1996) and yeast (Wodicka et al. 1997) gene expression and functional mapping of the yeast genome (Shoemaker et al. 1996). In each of these applications, oligonucleotide probes were selected and synthesized on the arrays as specific complements to each interrogated nucleotide in the targeted sequence. In this report, we have pursued the strategy of synthesizing an array that is composed of oligonucleotide probes specifically complementary to the M. tuberculosis rpoB gene and by use of this array as a generic tool to analyze the same gene from nine other nontuberculosis Mycobacterial species. The use of this array allowed for the simultaneous detection of mutations that confer rifampin resistance as well as species identification.
In the most straightforward use of this array, 41 M. tuberculosis isolates were observed to possess a total of 12 mutantrpoB alleles of involving 8 codons of the rpoB gene. Forty of the mutations were of the missense type and one mutation was a 6-base deletion. Three other M. tuberculosis isolates (TB10, TB15, TB76) were found to be phenotypically resistant but lacked any mutations within the sequenced 705 bp of the rpoB gene. Such isolates are not unexpected because ∼10% of isolates exhibit rifampin resistance through an unknown mechanism (Heym et al. 1994;Kapur et al. 1994; Williams et al. 1994).
Because rifampin resistance-conferring mutations in rpoB have also been observed in nontuberculous Mycobacteria species (Honoréand Cole 1993; Levin and Hatfull 1993; Musser 1995) and because the use of 16S genotyping has proven problematic in speciating some mycobacteria (Fox et al. 1992), the rpoB oligonucleotide array and conventional dideoxynucleotide sequencing were used to analyze sequence diversity within and between members of the mycobacterial genus. Among the 10 Mycobacterium species studied, analysis of the 705 nucleotides of the rpoB gene revealed that intraspecies variation was smallest within M. tuberculosis, M. xenopi, and M. kansasii species and greatest within M. gordonae, M. fortuitum, and M. smegmatisspecies. For some of these species, like M. gordonae, this result would not be unexpected as they are known to be a heterogeneous group. However, interspecies variation of the rpoB gene ranged considerably with M. avium and M. scrofulaceum,displaying the least, and M. chelonae and M. xenopidisplaying the greatest variation (Fig. 2). Importantly, the sequence analysis identified several species-specific single nucleotide polymorphisms among the ten Mycobacterium species. Hunt et al. (1994) described the presence of three M. tuberculosis-specific signature nucleotides while examining a 180-bp region of rpoB for resistance conferring mutations. In this study, we have identified many nucleotide positions for each of the nine nontuberculosis species, which when considered in a combinatorial fashion, provide a unique set of fingerprints for a particular Mycobacterium species.
When the DNA sequences of the rpoB and 16S genes were compared to measure the extent of allelic variation in each of 93 and 83Mycobacterium isolates (Table 3), respectively, very different perspectives of the genomic diversity within each species were obtained. For M. tuberculosis, M. scrofulaceum, M. cheilonae, and M. kansasii, allelic variation in both genes was similar and relatively low. Thus, the analysis of both genes provided similar indications of the diversity in each of the respective genomes. The observation of 3 rpoB alleles in a collection of 11 isolates of M. kansasii is consistent with the identification of five subspecies for these species (Picardeau et al. 1997). A similar representative picture was observed for the 16S andrpoB genes of M. avium, M. smegmatis, and M. fortuitum, although the overall allelic diversity was greater. However, analysis of the genetic profiles for M. xenopi, M. intracellulare, and M. gordonae yielded a different picture of genomic variability within each species. The allelic variation observed in M. xenopi by use of rpoB was low, whereas the variation in the 16S gene sequences for the 13 isolates studied in this species was higher. Conversely, the 16S sequence variation observed in isolates of M. intracellulareand M. gondonae indicated relatively lower levels of sequence diversity within these species, whereas rpoB sequences suggested higher levels of genetic diversity. When the DNA sequences of the rpoB or 16S gene sequences were used to cluster each of 81Mycobacterium isolates into species groups, by use of an unrooted neighbor joining distance matrix, 76 of the isolates were clustered into the same groups irrespective of which gene sequence was used. Of the five isolates (m4, m36, m48, m66, m125) that were clustered into different groups depending on which gene sequence was used, the sequences of the rpoB gene (m4, m48, m125) grouped three of the isolates in a manner similar to the grouping derived from the biochemical classifications performed by the contributing microbiological laboratories. Of the remaining isolates, two isolates (m36, m66) were placed in three different groupings.
These results emphasize several points. First, the identification of species on the basis of the sequence diversity present in 705 nucleotides of the rpoB gene was similar to species assignments derived from an analysis of 180 bp of 16S sequences. In a similar manner, Kapur et al. (1994), also provided evidence that polymorphisms present in the 65-kD heat shock protein gene could be used to identify mycobacterial species. Second, because several species of Mycobacterium exhibited an apparently large number of 16S and/or rpoB alleles, the use of single representative isolates for each species will not provide a sufficiently large database to accurately classify such isolates. A larger database of nucleotide sequences, derived from several regions of the genome, will be needed to determine species and subspecies identification accurately. Third, for those Mycobacterium species for which different genes provided different indications of the extent of genetic diversity, additional regions of the genome will need to be characterized. Those regions to be sequenced should reflect both rapidly and more slowly evolving regions. This approach would aid in both speciation/subspeciation groupings and individual isolate tracking.
Fox et al. (1992), made this point in their analysis of two Bacillus species, noting that use of 16S gene sequences for species identification is most useful in distinguishing relationships between genera and will resolve some species but not more recently diverged species. Additional sequence surveillance could be made coincident with analysis of genes in which mutations confer drug resistance. For example, genotypic analysis of the catalase-peroxidase gene (katG) (Heym et al. 1995; Musser et al. 1996) and the promoter region of the INH genes (Musser et al. 1996) for isoniazid resistance, ribosomal protein S12 gene (rspL) and the 16S rRNA genes (rrs) for streptomycin resistance (Fin et al. 1993; Sreevatsan et al. 1996), a subunit of DNA gyrase A gene (gyrA) for fluoroquinolone resistance (Musser 1995), pyrazinamidase/nicotinamidase gene (pncA) for pyrazinamide resistance (Scorpio et al. 1992;Scorpio and Zhang 1996; Sreevatsan et al. 1997) and the emboperon for ethamburtol resistance (Telenti et al. 1997) could all be used to simultaneously monitor drug resistance and provide data for species identification. An array interrogating many of these genes has been designed and synthesized recently and is currently being tested (Fig. 6).

A high-density oligonucleotide array used to genotype 731 bp ofrpoB, 2286 bp of katG, 356 bp of rpsL, 1683 bp of 16S, 731 bp of gyrA, 281 bp of inh orf, 341 bp of hsp 65 kd, 1097 bp of dnaJ, and 1279 bp of 32 Kdgenes. Additionally, specific insertion, deletions, and missense mutations inrpoB and katG are interrogated by the alternative allele-specific oligonucleotide probes at the bottom of the chip.
Finally, analysis of the hybridization patterns on the rpoBhigh-density oligonucleotide array provides another level of important information about the identity of the Mycobacterium isolates. It is striking that the grouping of the 121 Mycobacteriumisolates based on hybridization patterns was virtually identical to the clustering obtained by the dideoxynucleotide-based DNA sequence of therpoB gene. This result underscores two important conclusions: There are characteristic, conserved hybridization patterns for each of a species group and high-density arrays produce consistent results by use of different manufactured lots of arrays and multiple sample preparations.
These results follow in the footpath of other strategies used to identify Mycobacterium species on the basis of lipid profiles (Minnikinal and Goodfellow 1980; Butler et al. 1991; Lambert et al. 1996) or the fingerprint of isolates withMycobacterium-specific genetic elements (for review, see Small and vanEmbden 1994). The most definitive fingerprint approach would involve the interrogation of most, if not all, of the nucleotides of the Mycobacterium genome. Currently, the genomes of two isolates (H37RV and CSU#93) of M. tuberculosis are being sequenced. By use of the completed sequence as a template to develop a high-density array, nearly all of the nucleotides of the genome of other M. tuberculosis isolates, as well as isolates from otherMycobacterium species, could be analyzed. Pattern analysis algorithms like the ones presented in this study could be used to analyze the results of the genome-wide surveys to identify identical and divergent sequence regions. For M. tuberculosis isolates, regions with such sequence variations may be responsible for clinically or epidemiologically important phenotypes. For non-tuberculosis isolates, the hybridization patterns on a genome-wide level would permit grouping of isolates in a manner similar to that reported in this study. Furthermore, such a strategy holds the possibility that similar genomic arrays can be constructed for each of the major species of the eubacteria. These arrays could serve the dual role of surveillance of biologically/clinically important genomic regions (i.e., drug resistance, toxins, pathogenicity factors) as well as allow for direct analysis of the bacterial genome for identification and epidemiological purposes.
METHODS
Bacterial Isolates, Preparation of Genomic DNA, and Drug Sensitivity Measurements
Clinical and ATCC isolates of M. avium, M. chelonae, M. fortuitum, M. gordonae, M. kansasii, M. intracellulare, M. scrofulaceum, M. smegmatis, M. tuberculosis and M. xenopiwere grown on Lowenstein–Jensen slants or in BACTEC 13A broth (Becton–Dickinson. Sparks, MD) (Table 1) (Roberts et al. 1991). Species identification of each isolate used in this study in Table 1was originally performed at the center that contributed the sample. Methods used for species identification were the current biochemical, probe-based and microbacterial growth assays prevalent in each contributing group (Butler et al. 1991; Nolte et al. 1995). DNA obtained from most isolates were prepared by boiling one bacterial colony in 100 μl of water for 10 min (Kirschner et al. 1993;Vaneechoutte et al. 1993). Cellular debris was removed by brief centrifugation. For isolates obtained from Stanford University Medical Center, DNA was extracted as described elsewhere (vanEmbden et al. 1993). Drug susceptibility testing was performed with the proportion method with medium containing 1 μg/ml rifampin. The isolate was considered resistant if there was >1% growth on the rifampin containing medium as compared with the growth on the drug free media (Inderlied et al. 1995).
PCR Amplification and Molecular Cloning
A 705-bp segment of the rpoB gene from eachMycobacterium isolate was amplified by use of the following primer pair: rpoB-F (5′-CCCAGGACGTGGAGGCGATCACACCGCA-3′) and rpoB-R (5′-CGTCGCCGCGTCGATCGCCGCGC-3′). Amplification of a 1433-bp region of the 16S genes was accomplished with primers 16SF (5′- GTGCTTAACACATGCAAGTCGA-3′) and 16SR (5′-CAATCGCCGATCCCACCTT-3′). From the 100 μl boiled sample containing the Mycobacterium genomic DNA, 1–2 μl was amplified in a 100 μl PCR reaction. Each PCR reaction contained 200 nm of deoxynucleotide triphosphates, 200 nm of each primer, 2.5 units of Taq polymerase (Boehringer Mannheim, Indianapolis, IN), 10 mm Tris-HCl (pH 8.3), 50 mmKCl, 1.5 mm MgCl2, and 5% DMSO. PCR amplification reaction conditions consisted of an initial denaturation step of 5 min at 95°C, and 35 cycles consisting a denaturation step of 95°C for 1 min, primer hybridization at 60°C (68°C for M. tuberculosis) for 30 sec and polymerase extension at 72°C for 2 min. The PCR reaction was completed by primer extensions lasting for 10 min at 72°C. Unincorporated nucleotides and primers were removed by filtration through Microcon 100 columns (Amicon Inc, Beverly, MA). The 705-bp fragments of rpoB were cloned from the amplicon into a pT7/T3 α18 plasmid (GIBCO BRL, Gaithersberg, MD) by use ofBamHI and HindIII, linking into DH11S competentE. coli (Life Technologies, Gaithersberg, MD).
Nucleotide Sequencing Using High-Density Oligonucleotide Arrays and Dideoxynucleotide Methods
Dideoxynucleotide chain termination sequencing of therpoB and 16S genes (clones and PCR amplicons) was carried out on ABI instruments (models 373 and 377) by use of the cycle sequencing protocol recommended by the manufacturer (Perkin-Elmer Cetus, Foster City, CA). The primers used for the dideoxynucleotide sequencing of therpoB gene were T3 (5′-ATTAACCCTCACTAAAGGGA-3′), T7 (5′-TAATACGACTCACTATAGGG-3′), 105A (5′-GACCAGAACAACCCGC-3′), 105Z (5′-GCGGGTTGTTCTGGTC-3′), 276A (5′-GGCTCGCTGTCGGTGTA-3′), 276Z (5′-TACACCGACAGCGAGCC-3′), 372Z (5′-CGTCGGCGGTCAGGTA-3′), 377A (5′-GACCGCCGACGAGGAG-3′), 537A (5′-CAGATGGTGTCGGTGG-3′), and 537Z (5′-CACCGACACCATCTG-3′). The primers used for the sequence determination of 180 bp the 16S genes were 312Z (5′-GTCACCCCACCAACAAG-3′) and T3 (see above). Unincorporated dye terminators and primers were separated from the extension products by ethanol precipitation and the samples were dried in a vacuum centrifuge. The samples were resuspended in a loading buffer (5:1 deionized formamide/50 mm EDTA, at pH 8.0) and heat denatured at 90°C for 5 min prior to electrophoretic analysis with 6.0% and 4.25% polyacrylamide sequencing gels. The sequence data was edited and assembled by use of the Sequencher software package (Gene Codes Corp., Ann Arbor, MI). Distances between clusters of isolates were determined by use of Jukes–Cantor neighbor joining algorithm as part of the University of Wisconsin GCG software package (Jukes and Cantor 1989). The stability of the tree generated was verified by standard bootstrapping methods (Efron and Tibshirani 1993) consisting of 1000 cycles.
Fluorescently labeled RNA targets for chip-based sequencing were produced by a method similar to that described previously by Kozal et al.(1996). Briefly, PCR primers rpoB-F and rpoB-R were resynthesized to contain T3 or T7 promoter sequences. After PCR, fluorescein labeled RNA amplicons were generated by use of these primers in an in vitro transcription reaction with removal of unincorporated nucleotides accomplished by filtration through Microcon 100 columns. For each hybridization reaction ∼20 nm of the fluorescein-labeled RNA was fragmented in 30 mm MgCl2 at 95°C for 30 min to generate oligomeric-sized RNA fragments. The fragmented RNA was hybridized for 30 min at 22°C in a volume of 500 μl of 6× SSPE, 20% deionized formamide and 0.005% Triton X-100 by use of a fluidics station (Affymetrix, Santa Clara, CA). The high-density oligonucleotide array was then washed under high stringency in 1× SSPE, 20% deionized formamide, 0.005% Triton, followed by a short low stringency wash in 6× SSPE, 0.005% Triton. The chip was then analyzed with a confocal scanner (Affymetrix, Santa Clara, CA) at 11.25 μm/pixel resolution at 22°C. Data analysis, basecalling, and alignment of sequences was performed with GeneChip software (Affymetrix, Santa Clara, CA).
Pattern Discovery in Hybridization Images
Cluster analysis was used as an exploratory tool to examine whether each of the isolates could be grouped on the basis of the similarity of their hybridization patterns. For each isolate, the hybridization pattern was represented on an array of 5640 probe intensities (four probes per nucleotide, one perfect-match probe, and three mismatch probes). Each of the probe intensities is uniquely identified by its chip coordinates and, thus, can be correlated across each array. Two methods for determining the distance (dissimilarity) between pairs of isolates were used. The first method used only the 14102 perfect-matched probes and was based on linear regression analysis (Salvatore 1982; Hamilton 1992). The distance between two isolates was represented as (1 − r 2), wherer 2 is the square of the correlation coefficient between the matched probe intensities distributed over the 1410 perfect-matched probes. For this metric, similarity can be viewed as the extent to which the ranks of probe intensities is preserved between the two isolates. This measure is invariant to chip-wise linear transformations of the probe intensities. An alternative way to visualize this similarity measure is to consider that each of the two arrays of length n is a set of coordinates of a point inn-dimensional space (n = 1410). The correlation coefficient between the two arrays is the cosine of the angle formed between two vectors drawn from the origin to each of these points.
The second method uses all 5640 probe intensities without regard for which probes are complementary to the wild-type M. tuberculosis rpoB gene sequence. First, probe intensities for each isolate were standardized to a zero mean and unit variance by a normal score transformation (Blom 1958; Tukey 1962). The variance, over isolates, for each of the probe intensities was computed. Probe intensities that have little variability over isolates are, in most cases, not very informative about differences among the samples. Therefore, probe intensities with variances in the top 10% were retained (564 probe intensities). A principal components analysis on the 564 × 564 covariance matrix of the retained intensities was performed identifying 15 principal components that accounted for 93% of the observed variance among the 121 isolates. For each isolate, the corresponding 15 principal component scores were computed. The 15 principal component scores are mutually orthogonal. Each isolate is represented as a point in a 15-dimentional Euclidian space. The distance between two isolates was defined as the Euclidian distance between the two points in this space.
Each of the two methods produced a 121 × 121 interisolate distance matrix. These matrices were input into a hierarchical clustering procedure (20). The observations were clustered by use of four linkage methods: single, average, complete, and Ward’s minimum variance (Ward 1963). The clustering structures were similar for each of the four methods; the results for the single-linkage clustering is shown. The results of cluster analysis are visualized in two ways: as dendrograms (Johnson 1967) or as color grids derived by rearranging the rows and columns of the distance matrix to correspond to the obtained clustering structure and then displaying the pairwise distances with a color palette to represent the range of distances (noncorrelative = red to correlative = blue).
To assess the cohesion or stability of the resulting clusters, we performed a bootstrap analysis. For each cluster analysis, we ran 1000 bootstrap replications, where for each replication we resampled, with replacement, 121 observations from the original data set. Cluster analysis was performed on each replication. For each replication and for every cluster obtained in the original analysis, we computed the percentage of the observations that belonged to that cluster. The confidence values we report are these percentages averaged over the 1000 bootstraps.
Acknowledgments
We thank Drs. D. Lockhart, J. Warrington, and S. Fodor for helpful discussion and critical reading of the manuscript; G. Mamtora, A. Duong, and D. Dutta for providing technical assistance, B. Norvell, and T. Edwards, and V. Ebertz for designing graphics and preparing the manuscript. This research was supported by the National Institute of Allergy and Infectious Diseases (grant 1R43A140400) and by Affymetrix, Inc.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵7 Corresponding author.
-
E-MAIL tom_gingeras{at}affymetrix.com; FAX (408) 481-0422.
-
- Received December 10, 1997.
- Accepted February 17, 1998.
- Cold Spring Harbor Laboratory Press








