
Abstract
To accelerate gene discovery and facilitate genetic mapping in the protozoan parasite Toxoplasma gondii, we have generated >7000 new ESTs from the 5′ ends of randomly selected tachyzoite cDNAs. Comparison of the ESTs with the existing gene databases identified possible functions for more than 500 new T. gondiigenes by virtue of sequence motifs shared with conserved protein families, including factors involved in transcription, translation, protein secretion, signal transduction, cytoskeleton organization, and metabolism. Despite this success in identifying new genes, more than 50% of the ESTs correspond to genes of unknown function, reflecting the divergent evolutionary status of this parasite. A newly recognized class of genes was identified based on its similarity to sequences known only from other members of the same phylum, therefore identifying sequences that are apparently restricted to the Apicomplexa. Such genes may underlie pathways common to this group of medically important parasites, therefore identifying potential targets for intervention.
Toxoplasma gondii is a widespread opportunistic pathogen that causes disease in immunocompromised patients and in congenitally infected infants. T. gondii belongs to the phylum Apicomplexa, which consists primarily of obligate intracellular parasites that are phylogenetically ancient and related only distantly to model single-celled organisms (Gajadhar et al. 1991). The phylum Apicomplexa contains numerous parasites of medical and veterinary significance, including Plasmodium (the causative agent of malaria), Eimeria (the causative agent of coccidiosis in domestic animals), and Cryptosporidium (an opportunistic pathogen that causes diarrheal disease in animals and humans). T. gondii is unusual among this group of parasites in being amenable to both classical and molecular genetics (Roos et al. 1994; Boothroyd et al. 1995). Combined with the simplicity of in vitro propagation and production of mutants,T. gondii provides a powerful system for experimental studies. Despite these many attributes, it suffers from a lack of gene sequence information. The January 1996 edition of GenBank contained <30 protein-coding genes from T. gondii.
Single-pass sequencing of cDNAs to generate ESTs has become the fastest growing segment of the public DNA databases (Boguski et al. 1993). Although incomplete and error prone, ESTs remain a powerful means of gene discovery and for generating biologically informative probes for mapping studies (Adams et al. 1995; Hillier et al. 1996). The increased power of algorithms used to compare gene sequences and corresponding predicted amino acid sequences, such as BLAST (Altschul et al. 1990), have enabled identification of a significant number of homologs by comparison of EST sequences with more complete entries in the public genome databases. Such comparisons have been used to identify ancient conserved regions—sequences that are present across diverse phyla and that represent protein domains with highly conserved structures or functions (Green et al. 1993).
One arena where EST sequencing is likely to make its greatest impact is the discovery of genes from phylogenetically distant organisms. Such systems suffer from the dual hindrance of limited scientific study and a greater degree of sequence divergence from well-studied organisms, therefore limiting gene identification by heterologous probing. Such is the situation with many parasitic organisms, which, despite their medical importance, lag behind in the application of genetics to solving important biological problems.
The feasibility of EST analysis for gene discovery in T. gondii was demonstrated recently by a pilot project where ∼20% of ESTs identified putative homologs to known genes by conservative BLAST comparisons with the databases (Wan et al. 1995). Based on this initial success, we have undertaken a larger-scale project to sequence ∼10,000 randomly isolated cDNAs from the rapidly proliferating stage (called tachyzoite) of T. gondii. The results demonstrate that moderate-scale EST sequencing is a powerful means for identifying genes that may mediate important aspects of intracellular parasitism byT. gondii and related parasites.
RESULTS
EST Sequencing: Rapid and Efficient Identification
of New Genes
Single-pass sequencing from the 5′ end of 10,000 independent PCR-amplified T. gondii cDNAs was used to generate more than 7000 new ESTs that were submitted to GenBank. The use of semiautomated sequencing, processing, and annotation resulted in a total cost of <$15 per completed EST entry. Failures were primarily attributable to poor-quality sequence (2269 of 2676 or 85% of failures), whereas contaminants with vector, mitochondrial, or non-T. gondiisequences represented <5% of all failures. The remaining failures (∼10%) were attributable to sequences that were considered too short for useful analyses.
The average read of the good-quality sequence was 285 bp, sufficient to allow robust homology searches. The ESTs were searched against the nonredundant protein and nucleic acid database of GenBank using BLAST with various search parameters to create listings of putative genes with different match stringencies. Using a probability cut-off ofP ≤ 10−6, ∼30% (2183 out of 7165) of the ESTs were assigned putative identities at the time of submission. Following submission, homologies were listed for ∼45% of the ESTs by the National Center for Biotechnology Information (NCBI) using more permissive criteria (P ≤ 10−3). Both “putative assignments” designated by Washington University and “neighbors” assigned by NCBI can be viewed at the following web site:http://www2.ncbi.nlm.nih.gov/dbST/dbest_query.html. Adopting a more conservative cutoff (P ≤ 10−10), ∼27% of ESTs had a putative identity; these were compiled into a listing of 500 nonredundant homologs of existing genes (available athttp://www.ebi.ac.uk/parasites/toxo/toxpage.html). Although these putative homologs will require additional study to verify, it is apparent that EST sequencing is extremely cost-effective and has an excellent success rate for gene discovery.
Distribution of EST Clusters
ESTs were clustered by sequence similarity to reveal the number of times a given sequence was encountered. The majority of sequences occurred only a single time—58% of ME49 and 50% of RH sequences were singletons. Highly redundant sequences, those occurring >10 times, made up ∼34% of all successful reads. In total, 4062 unique sequences were identified by combining all singletons plus the number of unique clusters with two or more members. This number does not, however, provide a reliable estimate of the number of genes in T. gondii as the 5′ sequences used here were not anchored; therefore nonoverlapping or minimally overlapping sequences from the same gene result in separate clusters. The relatively low rate of redundancy encountered indicates that generation of normalized libraries designed to remove abundant mRNAs may not be necessary for an EST project of this scope.
The Most Abundant ESTs Are Similar in Both Libraries
To maximize the biological value of the data obtained, sequences were derived from two separate tachyzoite cDNA libraries from two commonly used strains that represent the clonal type I (RH) and type II (ME49) lineages (Sibley and Boothroyd 1992; Howe et al. 1996). A majority of the most abundant sequences correspond T. gondiigenes known previously that encode a variety of surface or secretory antigens (Table 1). Among the top 15 ESTs are a number of newly identified genes (no homology to current gene databases) that are prime candidates for further studies. Their relative abundances suggest that they encode proteins that have important roles in the biology of T. gondii. Whereas the frequencies of ESTs for most genes were similar between libraries, several abundant ESTs differed by three- to fivefold between the two libraries (Table 1; data not shown). When analyzed by comparative Northern blot, however, these differences proved not to be indicative of similar differences in mRNA abundances for reasons that are unknown (data not shown). The one exception was an EST family represented by the ME49 clone zy64e04 (dbEST id 571145) that detected a 6.3-kb mRNA in ME49 strain but not RH strain tachyzoites analyzed by Northern blot (data not shown). This gene occurs once in the RH strain ESTs, but is ∼14 times more abundant in ME49 strain ESTs (when corrected for the total number of sequences in each library).
Summary of Most Abundant ESTs by Library as Determined by Cluster Analysis
| Rank | RH tachyzoite cDNA library | ME49 tachyzoite cDNA library | ||||
| dbEST ID no. | no. | gene/description | dbEST ID no. | no. | gene/description | |
| 1 | 465887 | 107 | GRA1 p24 dense granule protein | 571382 | 42 | SAG1 p30 surface antigen |
| 2 | 466589 | 69 | GRA2 p28 dense granule protein | 571076 | 32 | GRA2 p28 dense granule protein |
| 3 | 465733 | 68 | GRA6 p32 dense granule protein | 571569 | 29 | GRA1 p24 dense granule protein |
| 4 | 465767 | 64 | SAG1 p30 surface antigen | 571208 | 28 | unknown (similar to RH 467174) |
| 5 | 465877 | 58 | GRA5 p21 dense granule protein | 571108 | 27 | SAG2 p22 surface protein |
| 6 | 466942 | 53 | SAG2 p22 surface antigen | 571574 | 17 | GRA5 p21 dense granule protein |
| 7 | 467174 | 34 | unknown (similar to ME49 571208) | 571542 | 15 | unknown (similar to RH 467213 |
| 8 | 465857 | 29 | S8 ribosomal protein | 571282 | 15[iii] | unknown (similar to RH 466613) |
| 9 | 466627 | 28 | GRA3 p30 dense granule antigen | 571237 | 11[iii] | unknown (similar to RH 466242) |
| 10 | 466406 | 26[ii] | unknown (similar to ME49 574388) | 571154 | 9 | GRA6 p32 dense granule protein |
| 11 | 465750 | 22 | unknown (similar to ME49 571377) | 571377 | 9 | unknown (similar to RH 465750) |
| 12 | 466392 | 21 | 35-kD toxoplasma polypeptide | 571392 | 9 | unknown (similar to Schizosaccharomyces pombe7.67961) |
| 13 | 467170 | 19 | unknown (similar to ME49 604952) | 571332 | 8 | S2 ribosomal protein |
| 14 | 465671 | 18 | S30 ribosomal protein | 571072 | 8 | L18a ribosomal protein |
| 15 | 466619 | 17 | S7–S8 ribosomal protein | 574528 | 8 | L22 ribosomal protein |
[i] Total number of ESTs for the RH tachyzoite cDNA library was 5359; total number of ESTs for the ME49 tachyzoite cDNA library was 1806.
[ii] EST frequency is fivefold more abundant in RH.
[iii] EST frequency is fivefold more abundant in ME49.
Analysis of the Quality of EST Data for Known Genes
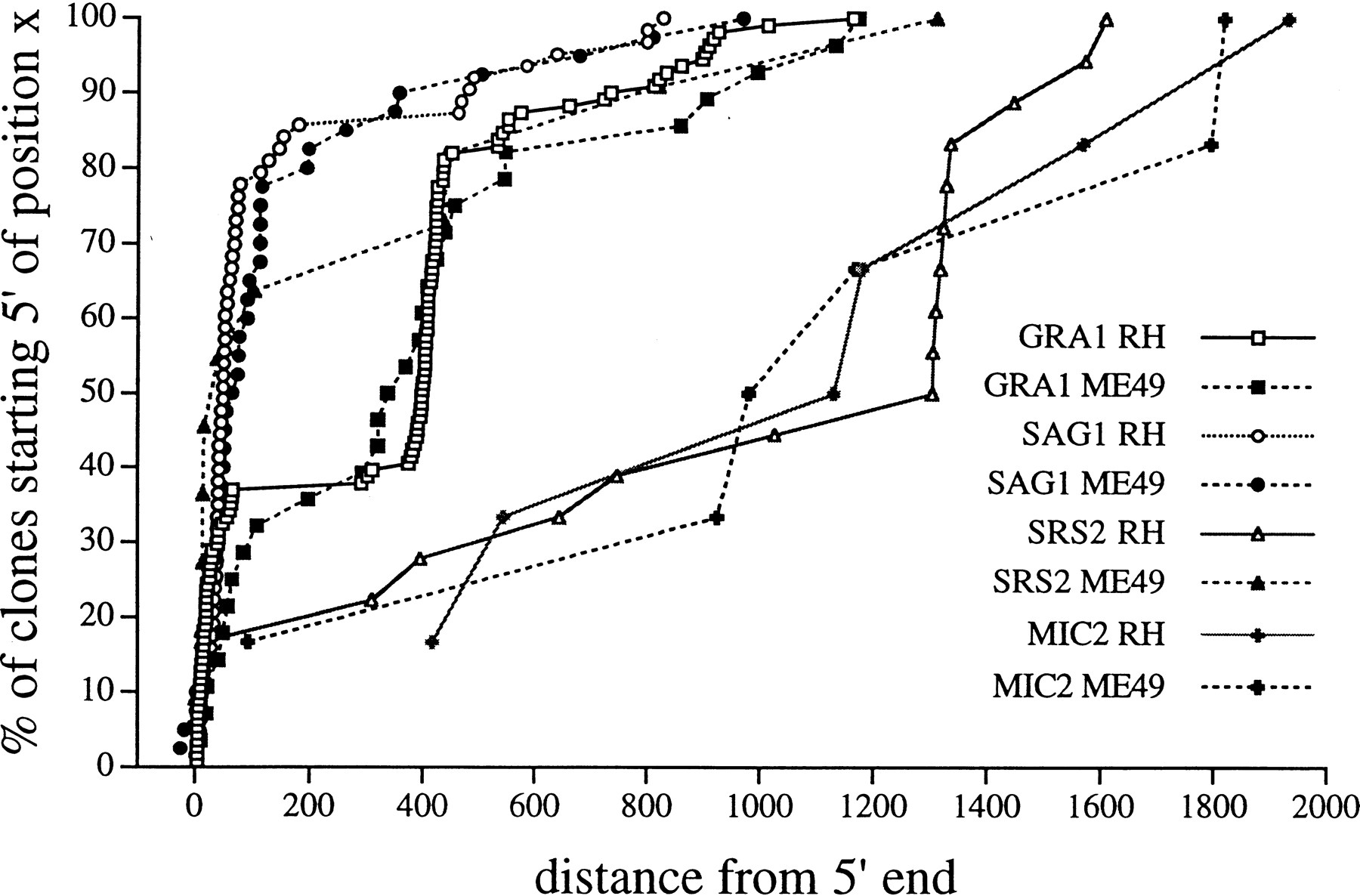
To establish the quality of sequence data generated from the two libraries, we analyzed the ESTs obtained for several well-studied, single-copy genes of T. gondii including MIC2(U62660), encoding a micronemal protein (Wan et al. 1997),GRA1 (M26007), encoding a calcium-binding dense granule protein (Cesbron-Delauw et al. 1989), and SAG1 (M23658), encoding the surface antigen p30 (Burg et al. 1988). The error rates for the high-quality portion of the ESTs corresponding to these genes were in the range of 1%–2%, consistent with previous reports of EST data quality (Hillier et al. 1996). To determine the distribution of sequences represented in the two libraries, the 5′ end of each EST corresponding to these genes was plotted. For SAG1 andGRA1, where the 5′ ends of transcripts are known (Burg et al. 1988; Cesbron-Delauw et al. 1989; Soldati and Boothroyd 1995), the EST sequences were strongly biased for the true 5′ ends (Fig. 1). The corresponding ESTs for the less abundant MIC2 gene and a newly described gene, SRS2, were scattered across their relevant transcripts (Fig. 1). In all three cases, multiple clusters were identified for each of the genes. ForMIC2, 13 separate ESTs were grouped into two distinct, nonoverlapping clusters that correspond to different regions of the same MIC2 gene. Such nonoverlapping clusters derived from the same gene also occurred in the more abundant GRA1 ESTs; however, they also defined a second allele defined by at least eight amino acid changes in a total of 191 residues that was only observed in the ME49 strain (data not shown).
Plot of the 5′ ends of ESTs for the genes GRA1, SAG1, MIC2, and SRS2. The majority of ESTs correspond to the extreme 5′ end of the transcripts reflecting the fact that the libraries contain primary full-length cDNAs. A strong premature stop is evident in GRA1, 400 bp downstream, and, in the RH library only, for SRS2 at 1300 bp downstream of the 5′-most ends. For the abundantly expressed genes, it is also possible to reconstruct an entire gene based on overlapping cDNAs.
Analysis of Abundant EST Clusters Reveals Gene Families
The nuclear genome of T. gondii is haploid and most previously characterized genes are single-copy, including theSAG1 gene. Multiple clusters for SAG1 occurred in both libraries because of the presence of similar, but distinct, members of a gene family. SAG1 encodes the major tachyzoite surface antigen p30 (Burg et al. 1988) that has distant homology toSAG3, which encodes a distinct surface antigen called p43 (Cesbron-Delauw et al. 1994). A second SAG1-related sequence (SRS1) has been identified recently by gene mapping experiments (Hehl et al. 1997). Analysis of the ESTs with homology toSAG1 reveals they define a family of at least six distinct genes as summarized in Table 2. In addition to the above described SRS1 gene, three new related genes were identified and given the names, SRS2, SRS3, and SRS4,respectively.
Identification of a Family of SAG1-Related Genes by EST Analysis
| Gene Product | Alias | Size (kD)[i] | Number of ESTs[ii] | Reference | GenBank accession no. |
| SAG1 | p30 | 32.9 | 106 | Burg et al. (1988) | M23658 |
| SAG3 | p43 | 41.8 | 10 | Cesbron DeLauw et al. (1994) | L21720 |
| SRS1 | — | 44.2 | 7 | Hehl et al. (1997) | U77677 |
| SRS2[iii] | — | 39.5 | 29 | this paper | N81803 |
| SRS3[iii] | — | 36.5 | 14 | this paper | N82063 |
| SRS4[iii] | — | ? | 1 | this paper | N81407 |
[i] Size of predicted primary translation product where complete ORF is known.
[ii] Number of essentially perfect matches in 7404 tachyzoite ESTs in dbEST.
[iii] SRS1 stands for SAG1-related surface antigens.
Classification of Conserved Protein Families
Comparison of the T. gondii ESTs with the P-fam database that contains 527 conserved protein domain families identified 80 nonribosomal homologs that fall into a number of different functional classes (Table 3). These homologies identify a large number of genes that have not been studied previously in T. gondii, therefore opening the way to direct analyses of their roles through molecular genetic studies. The absence of some conserved protein domains from the T. gondii ESTs is likely attributable in part to incomplete sampling of the genome (sequences obtained here come from only one of several developmental stages and are biased for highly expressed genes). Additionally, its phylogenetic position may account for an absence of families restricted to plants, archea or eubacteria and an absence of families related to metazoan specializations.
Functional Classification of Conserved Protein Domains vs. Yeast Proteins That Are Homologous to Toxoplasma ESTs
| Number of matching sequences[i] | |||
| P-fams | yeast proteins | ||
| Protein kinases/phosphatases | 2 | 4 | |
| RNA/DNA binding | 5 | 5 | |
| Translation/secretion | 3 | 18 | |
| Signaling | 4 | 3 | |
| Heat shock/chaperones | 7 | 11 | |
| Amino acid metabolism | 3 | 7 | |
| Detoxification | 4 | 3 | |
| Glycolysis/energy metabolism | 13 | 13 | |
| Nucleotide binding/ATP synthases | 6 | 6 | |
| Proteases | 3 | 3 | |
| Proteosome/ubiquitin | 3 | 11 | |
| GTPases/ATPases | 3 | 8 | |
| Cytoskeleton/cell adhesion/structural | 7 | 8 | |
| Ribosomal proteins | 25 | 67 | |
| Mitochondrial | 5 | 10 | |
| Purine/pyrimidine metabolism | 5 | 4 | |
| DNA/RNA replication/transcription | 7 | 20 | |
[i] Number of distinct conserved domain family members (P-fam) or yeast proteins recognized by toxoplasma ESTs.
The T. gondii ESTs were also compared with the yeast protein database derived from the recently completed genome sequencing project (Dujon 1996; Johnston 1996). Approximately 10% of T. gondiiESTs matched yeast ORFs (P ≤ 10−6). Excluding ribosomal protein-coding genes, 297 putative genes were identified by this comparison (listed on the T. gondii web page: http://www.ebi.ac.uk/parasites/toxo/toxpage.html). Homologs between the yeast protein database and T. gondii ESTs (P ≤ 10−10) were classified according to function and compared with the conserved protein families detected above (Table 3). Although similar numbers of homologs were detected for most categories, two functional classes stand out as being more common between yeast and T. gondii—translation/secretion and replication/transcription. This may reflect a greater similarity between T. gondii and yeast among these classes or simply be attributable to the greater knowledge of these factors within yeast relative to other taxa.
Identification of Sequences Restricted
to the Apicomplexa
Identification of genes that are restricted to the phylum Apicomplexa would be of considerable interest, both in biological terms and as potential targets for drug or vaccine design. We therefore searched dbEST and the nonredundant database of GenBank to identifyT. gondii ESTs exhibiting significant similarity to Apicomplexan sequences, while minimizing similarity to other taxa (Table 4). Several Apicomplexa-specific sequences were identified, including (1) secretory proteins associated with the specialized organelles that define this phylum (dense granules, rhoptries, micronemes) and apical antigens that are involved in adhesion (e.g., homologs of Pf-EMP1, AMA-1); and (2) genes that are expressed during specific developmental stages of the parasite life-cycle (e.g., homologs of trophozoite-, gametocyte-, and sporozoite-specific antigens in Eimeria and Plasmodium).
Phylogenetically Restricted Sequences of the Apicomplexa
| Reference (dbEST ID no.) | Contig ID[i] (no. of ESTs) | P value for apicomplexan genera | P value of closest nonapicomplexan | Putative identification | dbEST/ GenBank accession nos. |
| Apical antigens/adhesins | |||||
| 467174 | TQ-753 (92) | Neospora(10−22) | 100 | dense granule antigen | U72991 |
| 467174 | Plasmodium (10−3) | Pf-EMP1 | U27338 | ||
| 467347 | Sarcocystis(10−15) | 10−1 | microneme protein | L08892 | |
| 467347 | Eimeria(10−14) | A22655 | |||
| 466328 | TQ-702 (2) | Plasmodium(10−12) | 10−1 | apical membrane antigen I | L27503 |
| 475702 | TQ-1456 (2) | Plasmodium(10−14) | 10−10 | stippled structure-assoc. antigen | U10121 |
| Developmental antigens | |||||
| 466318 | TQ-11 (8) | Eimeria(10−50) | 10−1 | 19-kD sporozoite antigen | M59500 |
| 620297 | Plasmodium(10−13) | 10−7 | gametocyte-specific antigen Pfs97 | Z37724 | |
| 489378 | Plasmodium(10−11) | 10−1 | trophozoite antigen (Ser kinase?) | Z11832 | |
| 487888 | Plasmodium(10−11) | 10−6 | M83793 | ||
| 574122 | TQ-254 (12) | Eimeria(10−31) | 10−10 | developmental mRNA | M98840 |
| 574122 | Plasmodium(10−20) | M29000 | |||
| Metabolism, signaling, structural | |||||
| 513090 | Plasmodium(10−20) | 10−8 | RNA polymerase III (large subunit) | M73770 | |
| 653609 | TQ-1975 (4) | Plasmodium(10−13) | 10−9 | HMG protein | L31630 |
| 653609 | Babesia(10−13) | M81360 | |||
| 574303 | Plasmodium(10−11) | 10−4 | P-type ATPase | U39298 | |
| 574303 | Cryptosporidium(10−10) | U65981 | |||
| 487624 | Eimeria(10−23) | 10−14 | transhydrogenase | L08392 | |
| 467004 | Eimeria(10−15) | 10−6 | |||
| 489375 | Eimeria(10−11) | 10−2 | |||
| 488081 | TQ-1442 (11) | Neospora(10−28) | 10−4 | 14-3-3 regulatory protein | U31542 |
| 465739 | Neospora(10−23) | 10−16 | |||
| 571541 | Neospora(10−67) | 10−33 | |||
| Unknown function | |||||
| 467430 | TQ-627 (9) | Cryptosporidium(10−17) | 10−9 | EST (identity unknown) | AA224687 |
| 465760 | TQ-377 (4) | Plasmodium(10−39) | 10−10 | aspartate-rich protein | U46930 |
[i] Available from the Toxoqual database listing the consensus sequence for aligned EST contigs http:/daphne.humgen.upenn.edu/toxodb/
In addition to these Apicomplexa-specific genes, a number of ubiquitous enzymes and regulatory molecules were also identified by the above search criteria, including RNA polymerase III, HMG proteins, transhydrogenase, 14-3-3 proteins, etc. (Table 4). Based on their well-recognized functions, these would appear not to fit the criteria of phylogenetically restricted sequences. On closer examination, it was apparent these genes contain unique domains within otherwise highly conserved proteins. Therefore, although one or more ESTs matched only to other Apicomplexan sequences, other ESTs, corresponding to a different part of the same gene, were similar to a nonapicomplexan counterpart. Such comparative analysis provides a powerful means of identifying phylogenetically restricted domains within conserved proteins.
DISCUSSION
To accelerate gene discovery in T. gondii, we have generated >7000 ESTs from random cDNAs by rapid, semiautomatic single-pass sequencing. In total, more than 500 genes were newly identified in T. gondii based on BLAST comparisons with the DNA and protein databases with the conservative probability cut-off ofP ≤ 10−10. These genes fall into a number of families related to transcription, translation, protein secretion, and signaling. This rapid identification of new genes in T. gondii represents a 25-fold increase in the number of putative genes identified from this organism. In addition to providing evolutionary insight into an important group of parasites, these genes should foster studies on their roles in the biology of parasitism. Identification of phylogenetically restricted sequences of the Apicomplexa may also provide potential targets for intervention through chemotherapy or immunological means for the control of diseases caused by members of this phylum.
The use of ESTs to produce transcript maps has been heralded as a significant aid to positional cloning of genes, such as those involved in human genetic diseases (Adams et al. 1995). The ESTs generated here are being used currently to create such a genome map for T. gondii (J.W. Ajioka, C. Reitter, and R.M.R. Coulson, unpubl.) that will provide a framework for identification of genes involved in important biological phenotypes. Comparative EST analysis also provides a means for identifying strain polymorphisms (e.g., the GRA1example above) that may also be useful for genetic mapping studies.T. gondii has a complex life cycle that progresses from an acute stage, characterized by rapid proliferation of tachyzoites, to a slow-growing, encysted stage, the so-called bradyzoite, that is responsible for chronic infection. Further information on developmentally regulated genes should be forthcoming from a parallel effort to characterize a large number of ESTs from the bradyzoite stage of T. gondii (I.D. Manger, A. Hehl, S. Parmley, L.D. Sibley, M. Marra, L. Hillier, R. Waterston, and J.C. Boothroyd, in prep.).
T. gondii has an unusual and highly clonal population structure (Dardé et al. 1992; Sibley and Boothroyd 1992; Howe and Sibley 1995). The strain RH is the prototype for type I strains, which are all acutely virulent in mice (Sibley and Boothroyd 1992), whereas the type II strain ME49 is used commonly for models of chronic infection in animals (Hunter and Remington 1994). Type II strains are associated with the majority of human cases of toxoplasmosis both in AIDS and congenital infection (Dardé et al. 1992; Howe and Sibley 1995). One gene that has been implicated in pathogenesis isSAG1, which lies on chromosome VIII near a genetic locus for acute virulence in mice (Howe et al. 1996). Like SAG1, SRS1, SRS2, and SRS3 are expressed on the surface of tachyzoites as verified by antisera raised to the recombinant proteins (Hehl et al. 1997). Consequently, this family of proteins may be involved in mediating host cell interactions particularly as SAG1 has been implicated previously in cell adhesion (Kasper and Mineo 1994).
Identification of Conserved Protein Domains
in T. gondii
Highly conserved domains were identified by comparing T. gondii ESTs with the yeast protein database and to the P-fam listing of conserved protein families. These similarities identify a plethora of interesting genes for functional studies in T. gondii. Among the most abundant classes are genes with similarities to those involved in control of DNA replication/RNA transcription and protein translation and secretion. Developmental events are poorly understood in apicomplexan parasites, therefore, identification of genes involved in the machinery of replication and transcription should enable functional studies on the regulation of gene expression inT. gondii. Apicomplexan parasites are also highly specialized for regulated protein secretion (Carruthers and Sibley 1997). Despite the fact that yeast lacks such regulation, its secretion machinery is well understood. Identification of homologs involved in protein translation and secretion in T. gondii should allow studies on this important pathway in parasites. Given the enormous flexibility of genetic techniques and the functional information available about many genes in yeast, examination of these homologs may prove most fruitful using yeast as a heterologous system.
Identification of Sequences Restricted
to the Apicomplexa
The main features that define the phylum Apicomplexa are the presence of apically specialized organelles including a unique microtubule organizing center called the conoid (Russell and Burns 1984), a novel endosymbiotic organelle called the apicoplast (Kohler et al. 1997) and three classes of secretory organelles—micronemes, rhoptries, and dense granules (Aikawa and Sterling 1974). Sequential discharge of these secretory organelles accompanies invasion of T. gondii into the host cell suggesting that they perform distinct functions (Carruthers and Sibley 1997). Although some cytoskeletal antigens from apicomplexans are immunologically related (Taylor et al. 1990; Morrissette et al. 1994), secretory proteins typically show no cross-reactivity at the protein or nucleic acid levels, reflecting the deep phylogenetic branches within this phylum (Gajadhar et al. 1991). The discovery of proteins/genes in common to the Apicomplexa has been hindered by the labor-intensive nature of traditional comparative means.
To identify Apicomplexa-specific genes, we have exploited the recent expansion of the gene databases, including the growing collections of ESTs for T. gondii (this paper; Wan et al. 1995),Plasmodium falciparum (Reddy et al. 1993; Chakrabarti et al. 1994), and Cryptosporidium parvum (R. Nelson, unpubl.). Given that the unifying features of the phylum relate to apical specialization, it is perhaps not surprising that the phylogenetically restricted genes identified within the Apicomplexa include several proteins that may function in recognition and adhesion of host cells. For example, several T. gondii ESTs (including dbEST ID nos. 467174, 467408, and 466515) are similar to a conserved motif called the Duffy-binding-like (DBL) domain, which occurs in a variety of malarial proteins involved in red blood cell adhesion and in a family of malaria genes called var, the products of which mediate antigenic variation and sequestration of mature infected red cells (Su et al. 1995). This similarity occurs in the first of six motifs, a region defined by conserved cysteine and aromatic residues (Petterson et al. 1995; D. Petterson, pers. comm.) suggesting a common domain structure. Although T. gondii is not known to undergo either antigenic variation or sequestration, these DBL motifs may nevertheless identify proteins that participate in adhesion.
Other apicomplexan-specific genes encode developmentally regulated genes associated with the complex changes that occur during transformation of the life cycle stages. Finally, a number of unique metabolic genes were identified, including those involved in signaling and energy production. In some cases, these phylogenetically restricted domains were embedded within highly conserved proteins also found outside of the Apicomplexa. These two opposing definitions provide a powerful means for classifying protein similarities that span diverse phyla (e.g., ancient conserved regions) versus those restricted to a particular taxon. Expansion of the gene databases to include evolutionarily distant organisms will enable more refined analyses to identify phylogenetically restricted sequences, including those unique to the Apicomplexa. Application of similar searches in other organisms should likewise be a fruitful way to identify genes controlling biological traits that are of particular interest within specific taxa.
METHODS
Library Construction
Parasites were grown in human foreskin fibroblasts as described previously (Roos et al. 1994). Two separate laboratory strains were used for library construction—a clonal isolate of the RH strain and a clonal isolate of the ME49 strain called PDS. Freshly isolated tachyzoites were separated from host cell debris, washed in PBS, and RNAs were extracted with guanidium hydrochloride followed by phenol–chloroform treatment. cDNAs were synthesized from poly(A) mRNAs using the ZAP cDNA Synthesis Kit (Stratagene). The RH-strain library, referred to as “TgRH tachyzoite,” was described previously (Wan et al. 1995) and contains cDNAs directionally cloned into theEcoRI (5′ end) to XhoI (3′ end) sites of Lambda ZapII (Stratagene). A similar directional cDNA library, referred to as “TgME49 tachyzoite,” was constructed from PDS tachyzoite mRNAs.
Cycle Sequencing
Individual phage plaques grown on lawns of XL1 Blue strainEscherichia coli (Stratagene) were hand-picked and diluted in 20 μl of λ diluent in 96-well plates. The clones were each assigned an identification number consisting of TgESTzy or TgESTzz followed by a plate number (1–99), row letter, and column number. PCR amplifications were performed on the phage eluates using T3 and T7 primers to amplify the complete inserts in MJ Research PTC 200 thermal cyclers (Hillier et al. 1996). Amplification products were diluted in water and used as templates for 5′ sequencing reactions with Thermosequenase DNA polymerase and a DYEnamic ET T3-dye primer (Amersham) (Hillier et al. 1996). After thermal cycle sequencing, reactions were ethanol-precipitated, resuspended in loading buffer, and electrophoresed on ABI 373 sequencing machines.
Processing and Annotation
Following gel image analysis and DNA sequence extraction, ABI sequence data were automatically processed to (1) assess EST quality; (2) trim flanking vector sequences; (3) mask repetitive elements; (4) remove vector, bacterial, or human mitochondrial sequences; and (5) identify ribosomal RNAs and sequences highly similar to human mRNAs. The resulting sequences were annotated with similarity information, sequence quality information (i.e., position in the sequence at which high-quality data ends), library information, and submitted to dbEST/GenBank. The details of where these procedures differ from the previous ones are as follows (Hillier et al. 1996): As in (2), above the vector sequences were trimmed using the programs VEP (Dear and Staden 1991), WEP (W. Gish, unpubl.), and BLASTN2 (W. Gish, unpubl.) where S (score) = 133, S2 (minimum reported score) = 133, M (match) = 5, N (mismatch) = −11, W (word size) = 7, R (gap extension penalty) = 11, Q (gap initiation penalty) = 11, E2 (minimal reported e− value) = 0.5. WEP also served to identify incorrect adaptor sequences. As in (3), above, repetitive elements were identified and masked using blastx_and_mask (G. Miklem, unpubl.), which uses BLASTX (S = 50) to compare with a database of T. gondii repeat elements (GenBank accession nos.M57916, M57917, M57918, M57919, X60240, X60241, X60242, and X75429) translated in all six frames. The programs TANDEM and INVERTED (R. Durbin, unpubl.) were used to mask local tandem and inverted repeats, and DUST (R. Tatusov and D. Lipman, unpubl.) was used to mask low entropy sequence. As in (4), above, sequences determined to be vector (BLASTN2 S = 133, S2 = 133, M = 5 N = −11, W = 8 against a vector subset of GenBank), bacterial (BLASTN2 S = 133, S2 = 133, M = 5, N = −11 against the bacterial division of GenBank), or human mitochondrial (BLASTN2 S = 170, S2 = 150, M = 5, N = −11, R = 11, Q = 11 against GenBank:HUMMTCG, the human mitochondrion complete genome sequence) were not submitted to the public databases or included in further analysis. As in (5), above, EST similarities to ribosomal RNA were identified using BLASTN2 (S = 170, gapS2 = 150, M = 5, N = −11, R = 11, Q = 11) to compare with the RNA division of GenBank, resulting sequences were submitted to GenBank with the annotation of similar to ribosomal RNAs. EST similarities to human mRNAs were identified using BLASTN2 (S = 170, gapS2 = 150, M = 5, N = −11, R = 11, Q = 11) to search against a nonredundant human mRNA database (Boguski and Schuler 1995). These sequences were annotated as “similar to human mRNAs.” Fourteen ESTs were >95% identical to human mRNAs at the nucleotide level and these were annotated as “probable human contaminants.”
Identification of Gene Homologies
At Washington University, similarities to proteins were identified using BLASTX (M = PAM120, S = 100) searches against SWIR, which is a nonredundant protein database containing sequences culled from PIR, SWISS-PROT, and a database of predictedCaenorhabditis elegans proteins called WORMPEP (E. Sonnhamer, unpubl.). These identities were annotated at the time of entry and appear in the field “putative IDs” on the NCBI entries.
Following submission, T. gondii ESTs were compared with the NCBI nonredundant nucleotide database using BLASTN (W = 12, E2 = 10−6, the theoretical query size = 1000, theoretical database size = 100,000,000) and to the NCBI nonredundant protein database (August, 1996) using BLASTX (W = 3, matrix: BLOSUM62, T (threshold) = 11, two-hit window = 40 (two hits of threshold 11 must be found no farther than 40 apart), E2 = 0.01, theoretical query size = 300, theoretical database size = 20,000,000). The resulting top matches are listed in the “neighbors” field of the NCBI entries and are updated periodically.
Cluster Analysis for Grouping Similar ESTs
The ESTs were clustered using the program “est cluster” built around the icaass suite of tools (Parsons 1995). The algorithm compares each new EST longer than 100 bp with a growing set of representative ESTs clusters. Sequences were included with a cluster if the score was at least 50 (where matches are scored with +1 and mismatches are scored with −1). If the new sequence did not match an existing entry, then a new cluster was created. If the sequence was equivalent to more than one existing cluster, it was added to both.
EST Alignment and Construction of a Nonredundant Consensus Sequence Database
T. gondii EST sequences were also assembled into overlapping sequences using the “cap2” contig assembly program (Huang 1996) to align sequences with a high degree of sequence identity. Starting with 7869 quality EST sequences of >100 bp in length (includes all Toxoplasma ESTs in dbEST as of June 1997), 868 aligned clusters were identified, incorporating 4653 EST sequences. Each EST is represented only once in the aligned cluster database and a consensus sequence was determined for each contig defined (available athttp://daphne.humgen.upenn.edu/toxodb/toxodb.html). The entire “toxqual” data set (including consensus sequences and all unaligned ESTs) was compared with itself using BLASTN to identify similarities with P ≤ 10−10.
Analysis of Gene Families
Identification of sequences similar to SAG1 was performed by TBLASTN analysis of the dbEST database (NCBI server defaults; expect = 10, matrix = BLOSUM62) using coding sequences of the known SAGs [SAG1 (M23658), SAG3(L21720), and SRS1 (U77677); see Table 2] to identify matching ESTs. After exclusion of ESTs yielding low BLAST scores because of overall short or poor-quality sequence, ESTs were assigned to one of the known SAGs based on similarity. ESTs that did not match existing SAGs, were analyzed further by BLASTN and FASTA comparisons to dbEST and the resulting matches were assembled into families of overlapping sequences using GCG (I.D. Manger, A.B. Hehl, and J.C. Boothroyd, unpubl.).
Comparison of ESTs to Yeast Database
Each of the T. gondii ESTs was masked for low entropy sequence using DUST (R. Tatusov and D. Lipman, unpubl.) and compared with the 6218 yeast-translated ORFs/genes (Dujon 1996; Johnson 1996) using BLASTX with the following parameters: M = PAM120, T (word-score threshold) = 17, W = 4, V (no. of alignments reported) = 10000 filter seg. The yeast protein database was obtained from www-genome.stanford.edu(yeast_orfs.fasta).
Comparison to Conserved Protein Families
The 527 available seed alignments for protein families were obtained from release 2.0 of Pfam (Sonnhamer et al. 1997). Hidden Markov Models (HMM) were built using the hmmer-1.9j package with the following parameters: hmmb −d −R −PBLOSUM62. The −d option allows for maximum discrimination; the −R option allows that some family members may be fragmentary as occurs in EST data. The program HMMFS was then used to search the resulting HMMs against the entire set of ESTs, translated in all six frames using the program “orfer” (S. Eddy, unpubl.). A cutoff score of 20.0 bits was used to determine whether a given clone belonged to a specific family. This is a log-odds score; a score of 20 indicates that a sequence is 220-fold more likely to represent an authentic match than to occur by chance.
Identification of Phylogenetically Restricted Sequences
BLASTN and BLASTX searches of the nonredundant GenBank and dbEST databases were carried out using the GCG (v. 9.0 UNIX, December 1996) for each member of the consensus “toxqual” dataset and phylogenetic information was retrieved by matching the genus field for each match with P ≤ 10−10 with a look-up table (generated from the GSDB Taxonomy table) to assign a kingdom and phylum for each match. All entries in the toxqual dataset that showed no high probability match outside of the phylum Apicomplexa (P > 10−10 and at least 1000-fold greater than matches to apicomplexan sequences) were examined manually to facilitate annotation.
Financial support for the EST sequencing was provided by Merck Research Labs. Additional support for library construction and data analyses was provided by The Burroughs Wellcome Fund, The National Institutes of Health, Division of AIDS Research, The Wellcome Trust, and the Biotechnology and Biological Sciences Research Council. We thank Drs. Mervyn Turner (Merck Research Labs), Alex Fairfield, and Barbara Laughon (DAIDS) for their enthusiastic promotion of this project. We thank Sean Eddy, Dan Goldberg, and Mark Johnston for helpful comments and critical review of the manuscript. We gratefully acknowledge the assistance of Carolyn Tolstoshev and Jane Weisman at NCBI, the Washington University Genome Sequencing Center EST Team, and the University of Pennsylvania Computational Biology Program. The following individuals provided expert technical assistance: Nicole Dietrich, Tamara Kucaba, Maren Lingnau, John Martin, and Marinella Messina.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[9] Corresponding author.
Notes
[10] E-MAIL [email protected]; FAX (314) 362-1232.
REFERENCES
- ↵M.D. AdamsA.R. KerlavageR.D. FleischmannR.A. FuldnerC.J. BultN.H. LeeE.F. KirknessK.G. WeinstockJ.D. GocayneO. White(1995) Initial assesment of human gene diversity and expression patterns based on 83 million nucleotides of cDNA sequence. Nature 377:3–17.
- ↵M. AikawaC.R. Sterling(1974) Intracellular parasitic protozoa. (Academic Press, New York, NY).
- ↵S.F. AltschulW. GishW. MillerE.W. MyersD.J. Lipman(1990) Basic local alignment search tool. J. Mol. Biol. 215:403–410.
- ↵M. BoguskiG. Schuler(1995) Establishing a human transcript map. Nature Genetics 10:369–371.
- ↵M.S. BoguskiT.M.J. LoweC.M. Tolstoshev(1993) dbEST-database for “expressed sequence tags.”. Nature Genet. 4:332–333.
- ↵J.C. BoothroydM. BlackK. KimE.R. PfefferkronF. SeeberL.D. SibleyD. Soldati(1995) Forward and reverse genetics in the study of the obligate, intracellular parasite Toxoplasma gondii. Methods Mol. Genet. 6:3–29.
- ↵J.L. BurgD. PerlmanL.H. KasperP.L. WareJ.C. Boothroyd(1988) Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J. Immunol. 141:3584–3591.
- ↵V.B. CarruthersL.D. Sibley(1997) Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 73:114–123.
- ↵M.F. Cesbron-DelauwB. GuyR.J. PierceG. LenzenJ.Y. CesbronH. CharifP. LepageF. DarcyJ.P. LecocqA. Capron(1989) Molecular characterization of a 23-kilodalton major antigen secreted by Toxoplasma gondii. Proc. Natl. Acad. Sci. 86:7537–7541.
- ↵M.F. Cesbron-DelauwS. TomavoP. BeauchampsM.P. FourmauxD. CamusA. CapronJ.F. Dubremetz(1994) Similarities between the primary structures of two distinct major surface proteins of Toxoplasma gondii. J. Biol. Chem. 269:16217–16222.
- ↵D. ChakrabartiG.R. ReddyJ.B. DameE.C. AlmiraP.J. LapisR.J. FerlT.P. YangT.C. RoweS.M. Schuster(1994) Analysis of expressed sequence tags from Plasmodium falciparum. Mol. Biochem. Parasitol. 66:97–104.
- ↵M.L. DardéB. BouteilleM. Pestre-Alexandre(1992) Isoenzyme analysis of 35 Toxoplasma gondii isolates: Biological and epidemiological implications. J. Parasitol. 78:786–794.
- ↵S. DearR. Staden(1991) A sequence assembly and editing program for efficient management of large projects. Nucleic Acids Res. 19:3907–3911.
- ↵B. Dujon(1996) The yeast genome project: What did we learn. Trends Genet. 12:263–269.
- S. Eddy(1995) Multiple alignment using hidden markov models. Proceedings of the third international conference on intelligent systems for molecular biology (AAAI Press, Menlo Park, CA), pp 201–205.
- ↵A.A. GajadharW.C. MarquardtR. HallJ. GundersonE.V. Ariztia-CarmonaM.L. Sogin(1991) Ribosomal RNA sequences of Sarcocystis muris, Theileria annulata and Crypthecodinium cohnii reveal evolutionary relationships among apicomplexans, dinoflagelates, and ciliates. Mol. Biochem. Parasitol. 45:147–154.
- ↵P. GreenD. LipmanL. HillierR. WaterstonD. StatesJ.M. Claverie(1993) Ancient conserved regions in new gene sequences and the protein databases. Science 259:1711–1716.
- ↵A. HehlT. KreigerJ.C. Boothroyd(1997) Identification and characterization of SRS1, A Toxoplasma gondii surface antigen upstream of and related to SAG1. Mol. Biochem. Parasitol. 89:271–282.
- ↵L. HillierG. LennonM. BeckerM.F. BonaldoB. ChiapelliS. ChissoeN. DeitrichT. DubuqueA. FavelloW. Gish(1996) Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 6:807–828.
- ↵D.K. HoweL.D. Sibley(1995) Toxoplasma gondii comprises three clonal lineages: Correlation of parasite genotype with human disease. J. Infect. Dis. 172:1561–1566.
- ↵D.K. HoweB.C. SummersL.D. Sibley(1996) Acute virulence in mice is associated with markers on chromosome VIII in Toxoplasma gondii. Infect. Immun. 64:5193–5198.
- ↵X. Huang(1996) An improved sequence assembly program. Genomics 33:21–31.
- ↵C.A. HunterJ.S. Remington(1994) Immunopathogenesis of toxoplasmic encephalitis. J. Infect. Dis. 170:1057–1067.
- ↵M. Johnston(1996) The complete code for a eukaryotic cell. Curr. Biol. 6:500–503.
- ↵L.H. KasperJ.R. Mineo(1994) Attachment and invasion of host cells by Toxoplasma gondii. Parasitol. Today 10:184–188.
- ↵S. KohlerC.F. DelwicheP.W. DennyL.G. TilneyP. WebsterR.J.M. WilsonJ.D. PalmerD.S. Roos(1997) A plastid of probable green algal origin in Apicomplexan parasites. Science 275:1485–1489.
- ↵N.S. MorrissetteV. BedianP. WebsterD.S. Roos(1994) Characterization of extreme apical antigens from Toxoplasma gondii. Exp. Parasitol. 79:445–459.
- ↵J. Parsons(1995) Improved tools for DNA comparison and clustering. Comp. Appl. Biosci. 11:603–613.
- ↵D.S. PettersonL.H. MillerT.E. Wellems(1995) Isolation of multiple sequences from the Plasmodium falciparum genome that encode conserved domains homologous to those in erythrocyte-binding proteins. Proc. Natl. Acad. Sci. 92:7100–7104.
- ↵G.R. ReddyD. ChakrabartiS.M. SchusterR.J. FerlE.C. AlmiraJ.B. Dame(1993) Gene sequence tags from Plasmodium falciparum genomic fragments prepared by the “genase” activity of mung bean nuclease. Proc. Natl. Acad. Sci. 90:9867–9871.
- ↵D.S. RoosR.G.K. DonaldN.S. MorrissetteA.L. Moulton(1994) Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol. 45:28–61.
- ↵D.G. RussellR.G. Burns(1984) The polar ring of coccidian sporozoites: A unique microtubule-organizing centre. J. Cell Sci. 65:193–207.
- ↵L.D. SibleyJ.C. Boothroyd(1992) Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359:82–85.
- ↵D. SoldatiJ.C. Boothroyd(1995) A selector of transcription initiation in the protozoan parasite Toxoplasma gondii. Mol. Cell. Biol. 15:87–93.
- ↵E.L.L. SonnhamerS.R. EddyR. Durbin(1997) Pfam: A conprehensive database of protein families based on seed alignments. Proteins 28:405–420.
- ↵X. SuV.M. HeatwoleS.P. WertheimerF. GuinetJ.A. HerrfeldtD.S. PetersonJ.A. RavetchT.E. Wellems(1995) The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 82:89–100.
- ↵D.W. TaylorC.B. EvansS.B. AleyJ.R. BartaH.D. Danforth(1990) Identification of an apically-located antigen that is conserved in sporozoan parasites. J. Protozool. 37:540–545.
- ↵K.L. WanJ.M. BlackwellJ.W. Ajioka(1995) Toxoplasma gondii expressed sequence tags (ESTs): Insight into tachyzoite gene expression. Mol. Biochem. Parasitol. 84:203–214.
- ↵K.L. WanV.B. CarruthersL.D. SibleyJ.W. Ajioka(1997) Molecular characterisation of an expressed sequence tag locus of Toxoplasma gondii encoding the micronemal protein MIC2. Mol. Biochem. Parasitol. 84:203–214.